Futibatinib: An Oral Irreversible FGFR1-4 Inhibitor
TAS-120 (futibatinib)
oral irreversible FGFR1-4 inhibitor in Ph. I-III trials for FGFR+ advanced tumors from structure-based design Cancer Res., Sep. 24, 2020 Taiho Pharmaceutical, Tokyo, JP

Other molecules you may be interested in
revumenib
Revumenib (Revuforj®) is approved by the FDA as an oral, first-in-class menin-MLL interaction inhibitor for acute leukemia (AL). Since inhibition of the menin-MLL fusion protein interaction is selective for AL with a KMT2A translocation, it does not compromise normal hematopoietic function. This article covers the background, optimization and clinical development that has led to the approval of this groundbreaking new drug in a hard-to-treat indication.


RMC-6236
RMC-6236 is a non-covalent pan-RAS(ON) inhibitor from Revolution Medicines, which shows remarkable efficacy in tumors driven by RAS mutants that were previously considered “undruggable,” such as G12V/D/A/S, G13X, and Q61X. RMC-6236 exerts its action via a “tri-complex” mechanism, gluing RAS to the ubiquitously expressed chaperone protein, cyclophilin A. Our in-depth article covers the presentation given at the AACR 2024 meeting, which outlines the discovery and preclinical profile of RMC-6236 as well as the latest clinical updates.

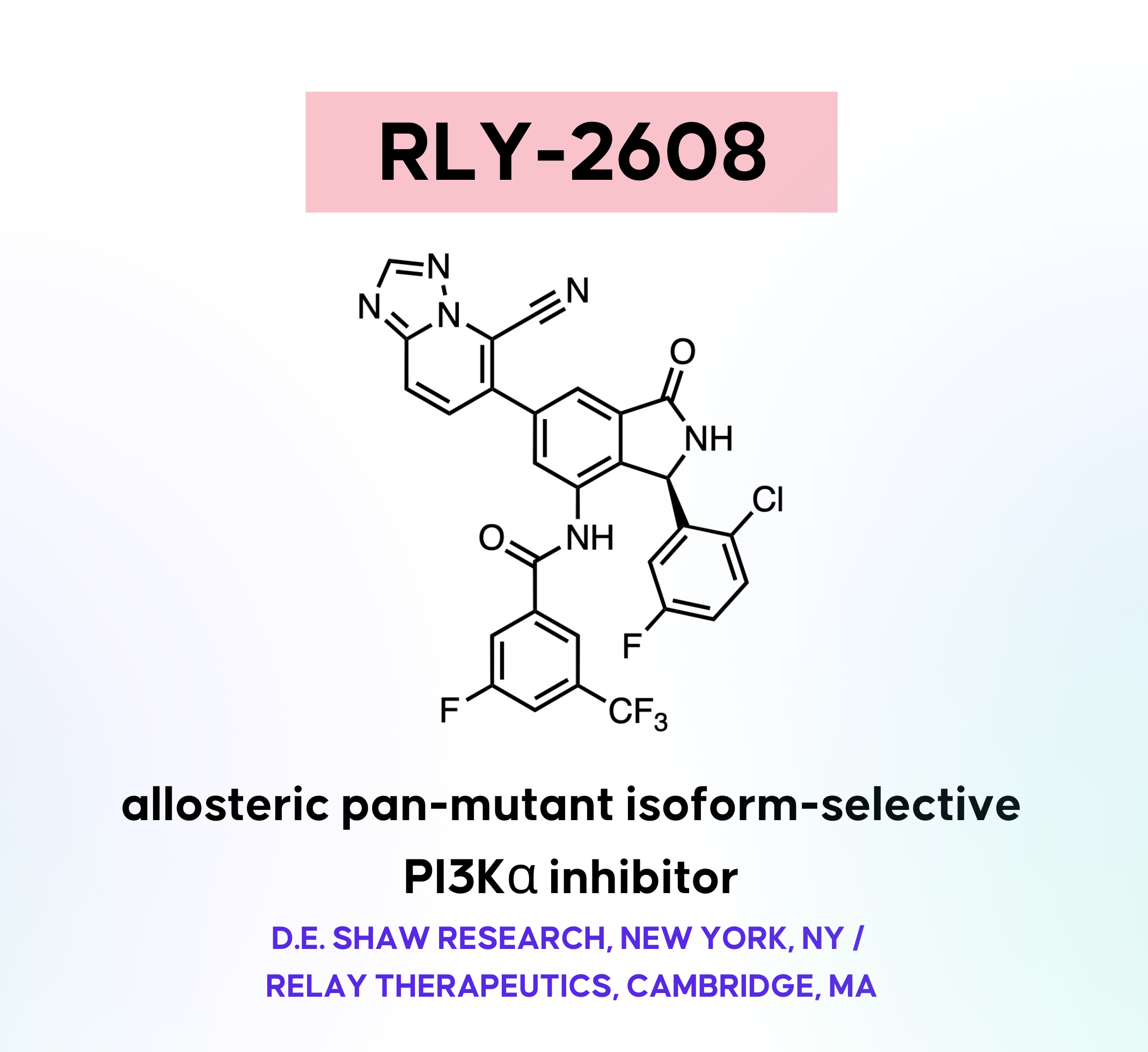
RLY-2608
RLY-2608 is an oral, mutant-selective PI3Kα allosteric inhibitor from Relay Therapeutics. Current FDA-approved PI3Kα modulator (alpelisib) and a clinically advanced molecule (inavolisib) are limited by their off-target toxicities associated with the inhibition of WT PI3Kα, leading to hyperglycemia and rash. RLY-2608 is currently in a Ph. I as a single agent and in combination with fulvestrant for HR+/HER2- breast cancer treatment. This article reviews the discovery of RLY-2608, its mechanism of mutant selectivity, how it compares to other molecules, recent clinical developments, and more.
AZ-PRMT5i-1
AZ-PRMT5i-1 is an orally bioavailable MTA-cooperative PRMT5 inhibitor that specifically targets MTAP-deleted cancers and is structurally related to AZ’s clinical candidate, AZ3470. This case study is an excellent example of utilizing bioisosteric replacements for polar guanidine headgroups, rigidifying scaffolds through spirocyclization to reduce rotatable bonds, and leveraging fluorine atoms beyond simply blocking metabolic soft spots.

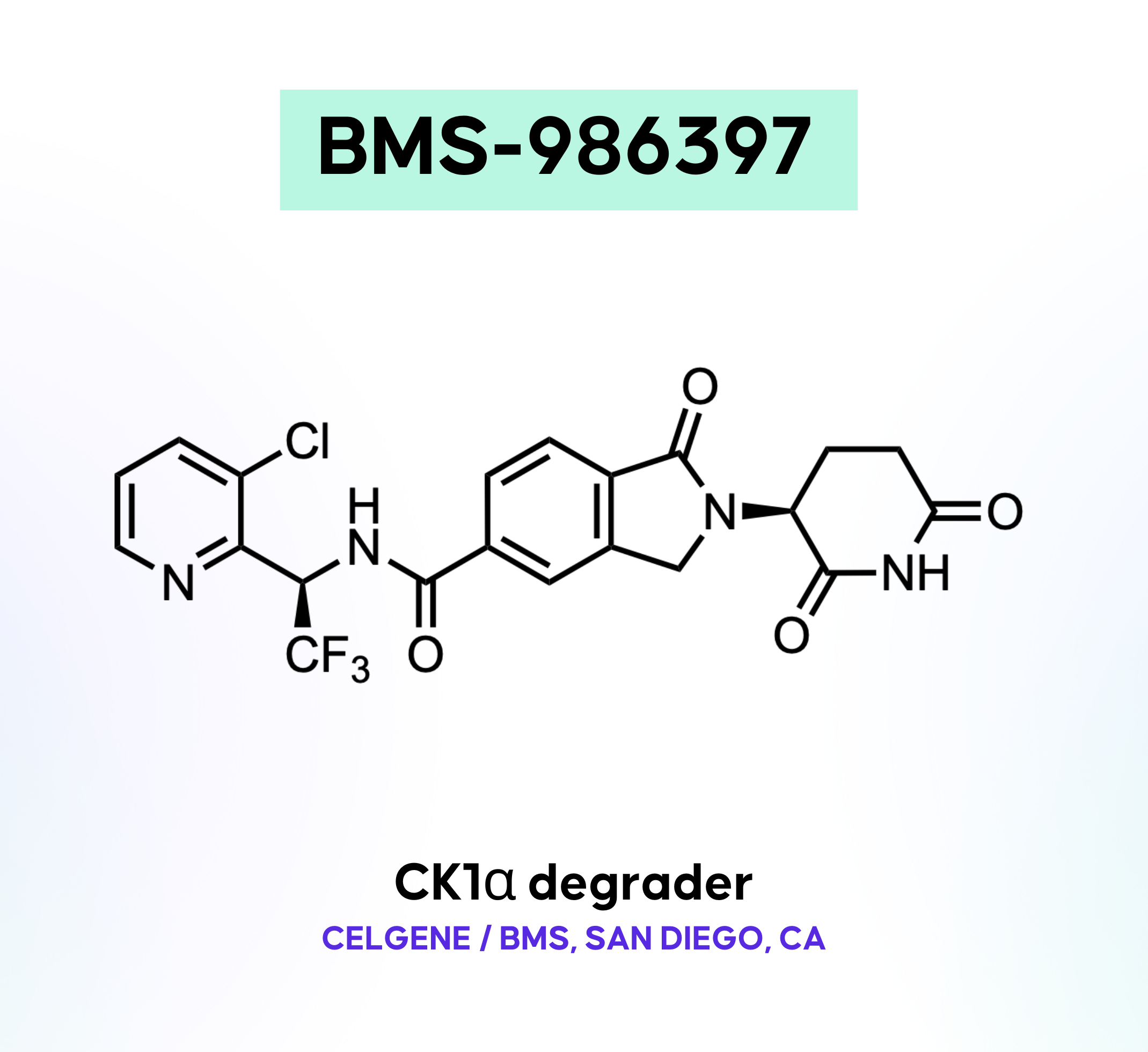
BMS-986397
BMS-986397 is a potential first-in-class CRBN-based selective CK1α molecular glue degrader. CK1α promotes tumor growth by enhancing MDM2 and MDMX degradation of the tumor suppressor p53. Since AML has a low TP53 mutation rate, activating the p53 pathway is a promising approach; however, p53 activators have faced challenges due to hematological toxicities. Targeting CK1α degradation offers an alternative approach. The BMS team sought to develop a CELMoD® for CK1α degradation. This article outlines the discovery of BMS-986397, as presented at the ACS Fall 2024 meeting in Denver, CO.