A Next-Generation, Mutant-Selective PI3Kα Allosteric Inhibitor with a Potentially Improved Safety Profile
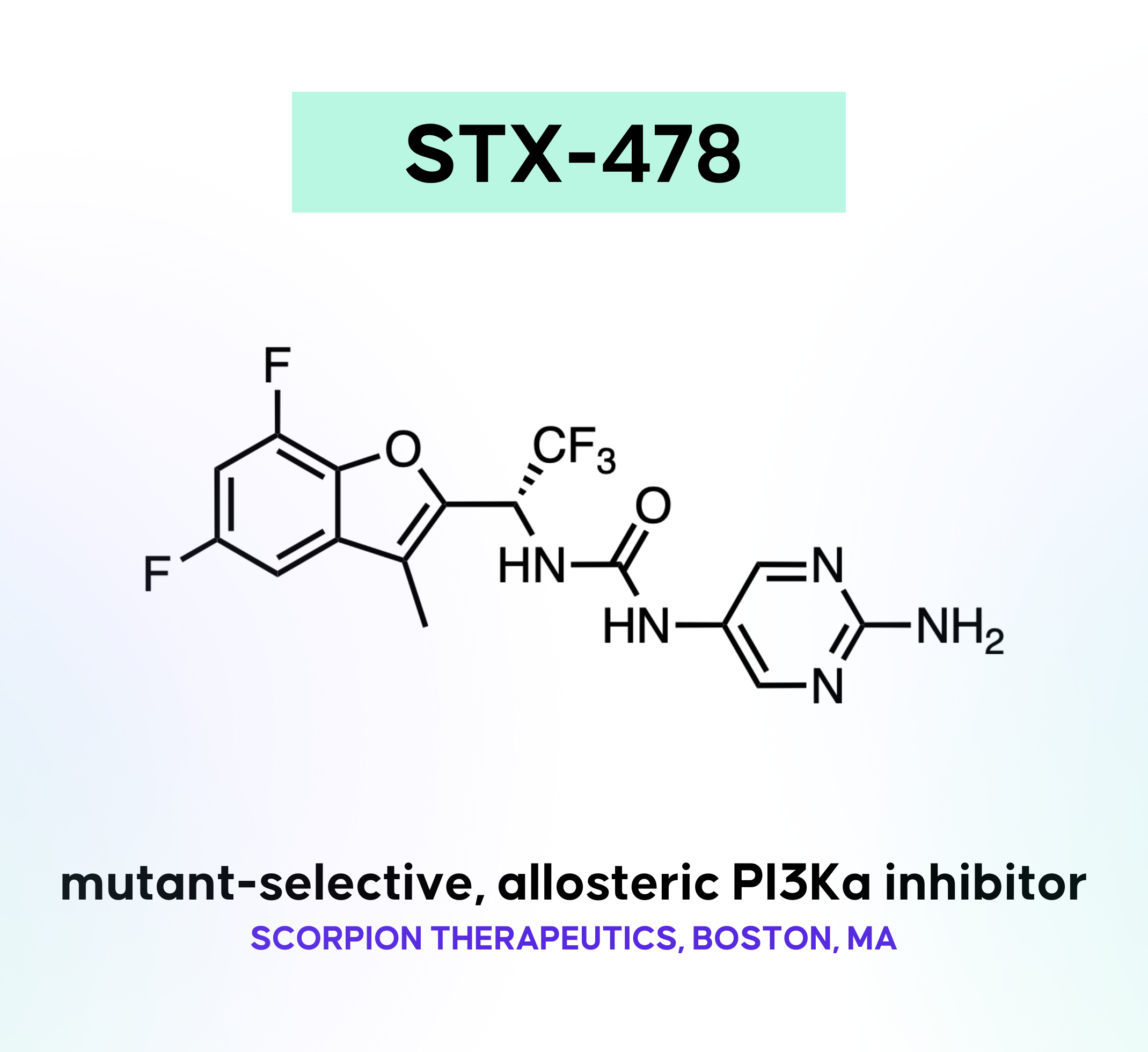
STX-478
mutant-selective, allosteric PI3Ka inhibitor Ph. I/II for breast cancer and other solid tumors from ASMS (affinity selection mass spectrometry) screen Cancer Discov., August 2023 Scorpion Therapeutics, Boston, MA

Other molecules you may be interested in
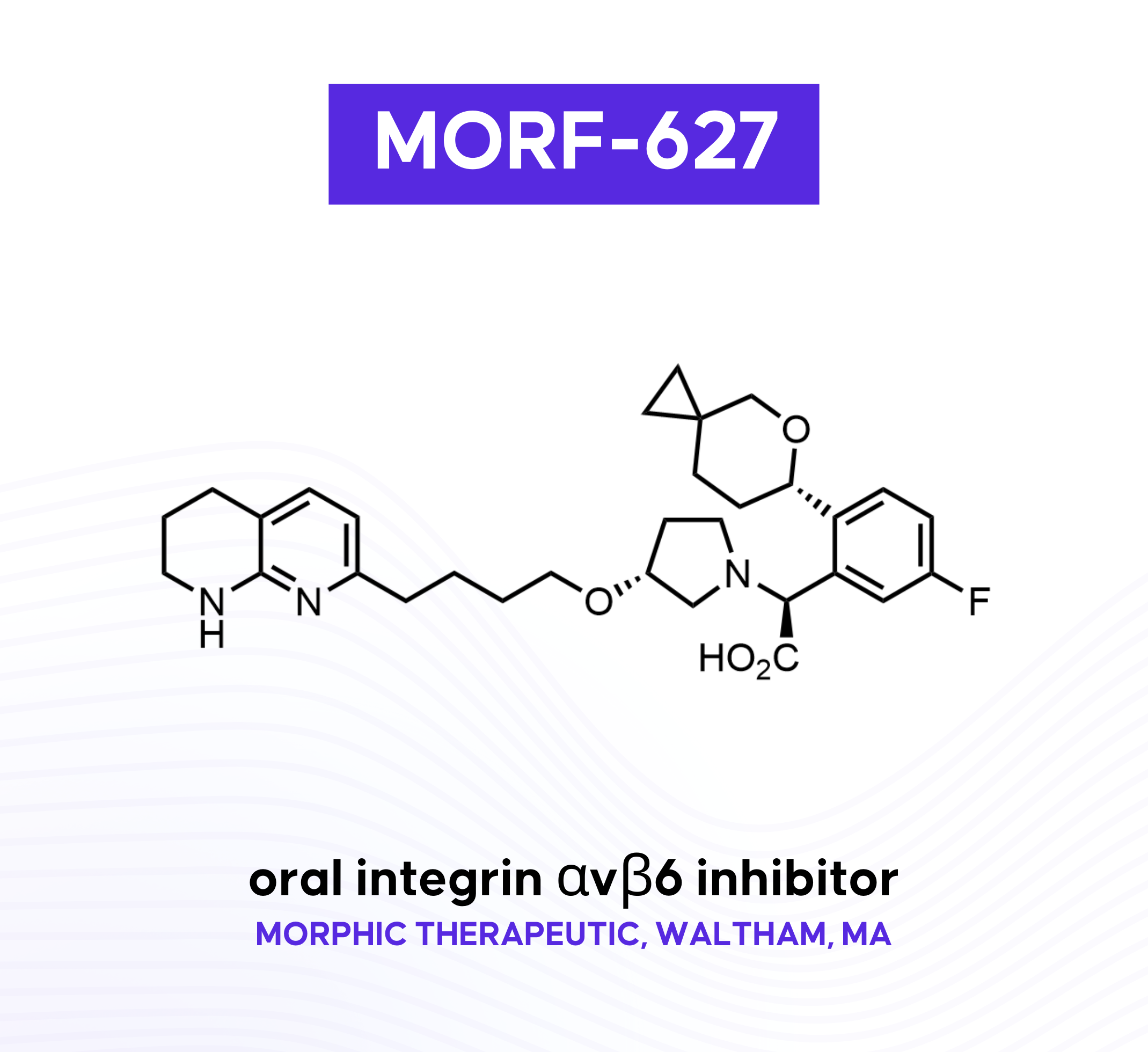
MORF-627
Morphic Therapeutic’s MORF-627 is an oral αvβ6 integrin inhibitor designed to treat IPF by blocking the TGFβ pathway. The Morphic team leveraged SBDD and FEP+ during lead optimization to enhance permeability and isoform selectivity. This article highlights the team’s focus on inhibiting the “bent-closed” conformation of αvβ6 as well as the linker design that led to the “chameloenicity” of the lead compound. The impressive multispecies PK and in vivo efficacy of MORF-627 in preclinical models was unfortunately accompanied by oncogenic toxicity that prevented it from reaching clinical trials.
inavolisib
Inavolisib is a PI3Kα isoform-selective kinase inhibitor and monovalent degrader of the mutant p110α catalytic subunit of mutant PI3Kα. The molecule selectively depletes mutant p110α in cancer cells with active RTK (receptor tyrosine kinase) signaling and is in several ongoing or planned Ph. III trials for breast cancer. In October 2024, it received FDA approval for use in combination with palbociclib and fulvestrant to treat adults with endocrine-resistant, PIK3CA-mutated, HR+/HER2- breast cancer. This article explains how it works, how it was discovered, and why it matters.

ABBV-467
AbbVie’s ABBV-467 is a highly potent, selective MCL-1 (myeloid cell leukemia-1) inhibitor which entered the clinic in 2022 in a Ph. I trial in patients with advanced hematologic cancers. MCL-1 has had a rough time in the clinic with multiple trials being halted or terminated due to cardiac toxicity, which is suspected to be an on-target effect. AbbVie’s approach with ABBV-467 was to target a highly potent/short half-life compound which could induce rapid apoptosis and tumor regressions in a short exposure period before the onset of any adverse events. Is this the end for MCL-1?

HC-7366
HiberCell recently disclosed the discovery of HC-7366, a potential first-in-class, intentionally discovered, orally bioavailable, potent, selective, small-molecule kinase activator of GCN2. HC-7366 has now progressed to Ph. I trials to treat ccRCC and AML. This case study is a fantastic example of how to mitigate CYP3A4 inhibition and improve oral bioavailability via judicious choice of salt formulation.

NDI-101150
NDI-101150 is an oral HPK1 inhibitor discovered by Nimbus Therapeutics and is currently in Ph. I/II clinical trial in advanced solid tumors. HPK1 is a compelling immuno-oncology target due to its critical role in regulating T-cells, B-cells, and dendritic cell-mediated immune responses. HPK1-deficient mice demonstrate enhanced anti-tumor T-cell responses and resistance to tumor growth. In this article, we detail the discovery of NDI-101150, as highlighted by Nimbus at the ACS Fall 2024 First-Time Disclosures session, interim results from the clinic, and more.
