A diaziridine-based tumor-activated prodrug for AKR1C3-expressing cancers from a defunct Bay Area biotech.
OBI-3424
IV AKR1C3-activated cytotoxin prodrug Ph. I/II in advanced solid tumors prodrug of DNA cross-linking agent Br. J. Cancer, May 12, 2023 OBI Pharma, TW; Threshold Pharma, CA

Other molecules you may be interested in
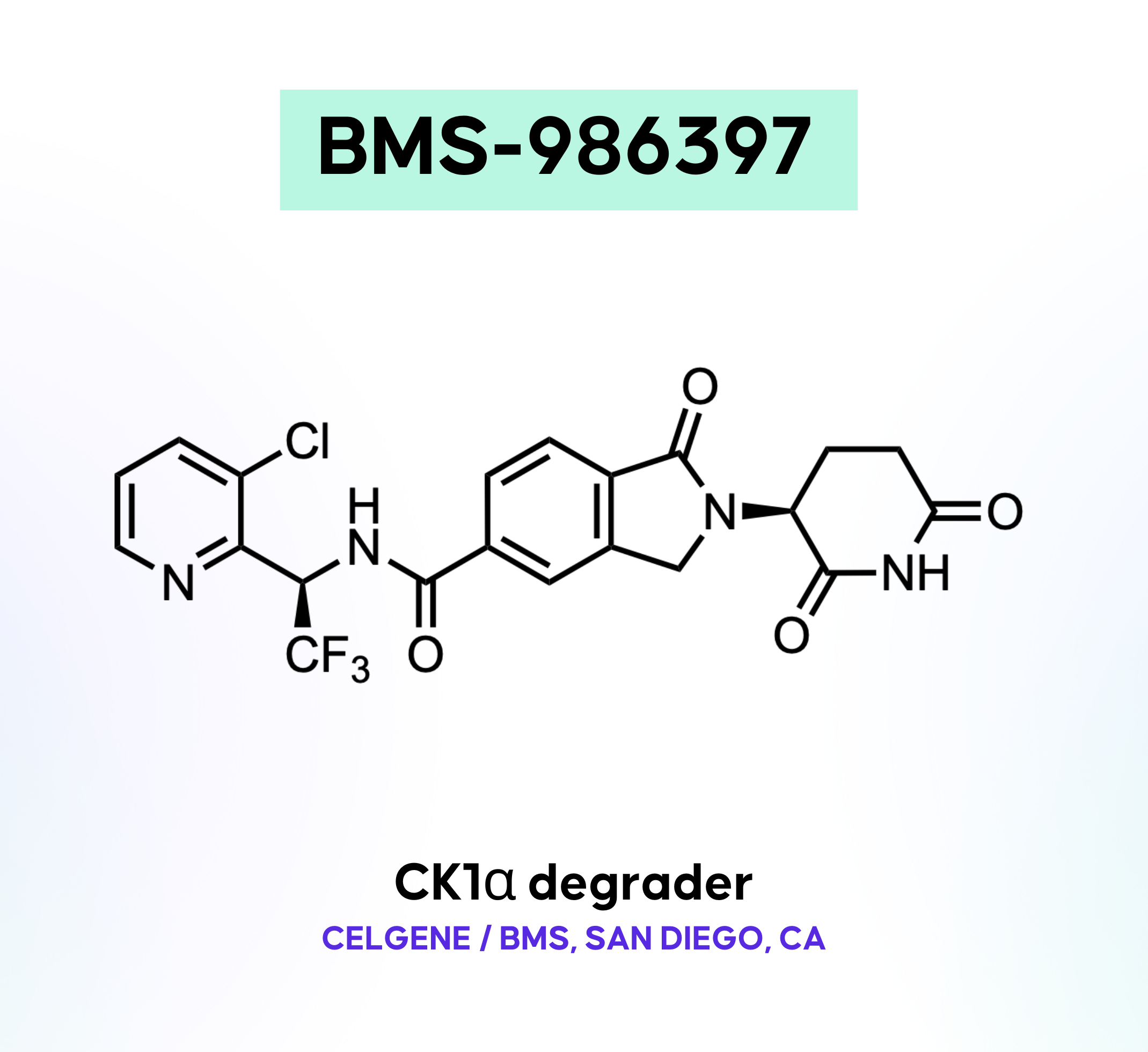
BMS-986397
BMS-986397 is a potential first-in-class CRBN-based selective CK1α molecular glue degrader. CK1α promotes tumor growth by enhancing MDM2 and MDMX degradation of the tumor suppressor p53. Since AML has a low TP53 mutation rate, activating the p53 pathway is a promising approach; however, p53 activators have faced challenges due to hematological toxicities. Targeting CK1α degradation offers an alternative approach. The BMS team sought to develop a CELMoD® for CK1α degradation. This article outlines the discovery of BMS-986397, as presented at the ACS Fall 2024 meeting in Denver, CO.
AZ-PRMT5i-1
AZ-PRMT5i-1 is an orally bioavailable MTA-cooperative PRMT5 inhibitor that specifically targets MTAP-deleted cancers and is structurally related to AZ’s clinical candidate, AZ3470. This case study is an excellent example of utilizing bioisosteric replacements for polar guanidine headgroups, rigidifying scaffolds through spirocyclization to reduce rotatable bonds, and leveraging fluorine atoms beyond simply blocking metabolic soft spots.

inavolisib
Inavolisib is a PI3Kα isoform-selective kinase inhibitor and monovalent degrader of the mutant p110α catalytic subunit of mutant PI3Kα. The molecule selectively depletes mutant p110α in cancer cells with active RTK (receptor tyrosine kinase) signaling and is in several ongoing or planned Ph. III trials for breast cancer. In October 2024, it received FDA approval for use in combination with palbociclib and fulvestrant to treat adults with endocrine-resistant, PIK3CA-mutated, HR+/HER2- breast cancer. This article explains how it works, how it was discovered, and why it matters.

BMS-986408
BMS-986408 is an oral, dual DGK ⍺ and ζ inhibitor currently in a Ph. I/II trial in patients with solid tumors. The compound was identified stemming from a phenotypic screening approach, and subsequent target deconvolution revealed DGK to be the target. If approved, it would be a first-in-class intracellular checkpoint inhibitor of DGK. This is an excellent case study on how to overcome a DILI risk associated with BSEP inhibition, as well as how to improve solubility and PK properties of your compounds through the introduction of polarity, reduction of aromatic rings, and increase in f(sp3).

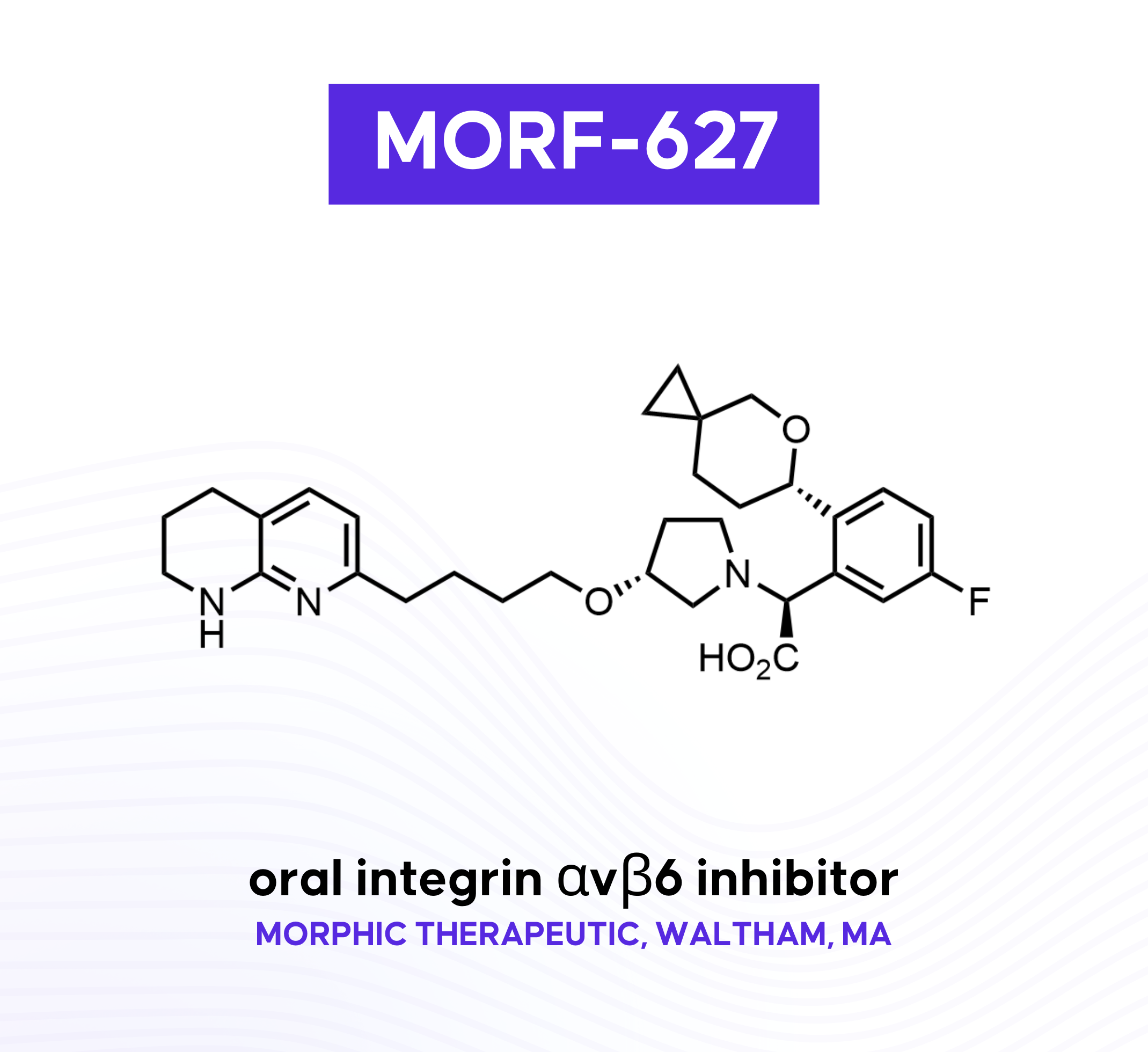
MORF-627
Morphic Therapeutic’s MORF-627 is an oral αvβ6 integrin inhibitor designed to treat IPF by blocking the TGFβ pathway. The Morphic team leveraged SBDD and FEP+ during lead optimization to enhance permeability and isoform selectivity. This article highlights the team’s focus on inhibiting the “bent-closed” conformation of αvβ6 as well as the linker design that led to the “chameloenicity” of the lead compound. The impressive multispecies PK and in vivo efficacy of MORF-627 in preclinical models was unfortunately accompanied by oncogenic toxicity that prevented it from reaching clinical trials.