First Disclosures of Small Molecule Drug Candidates – EFMC-ISMC 2021
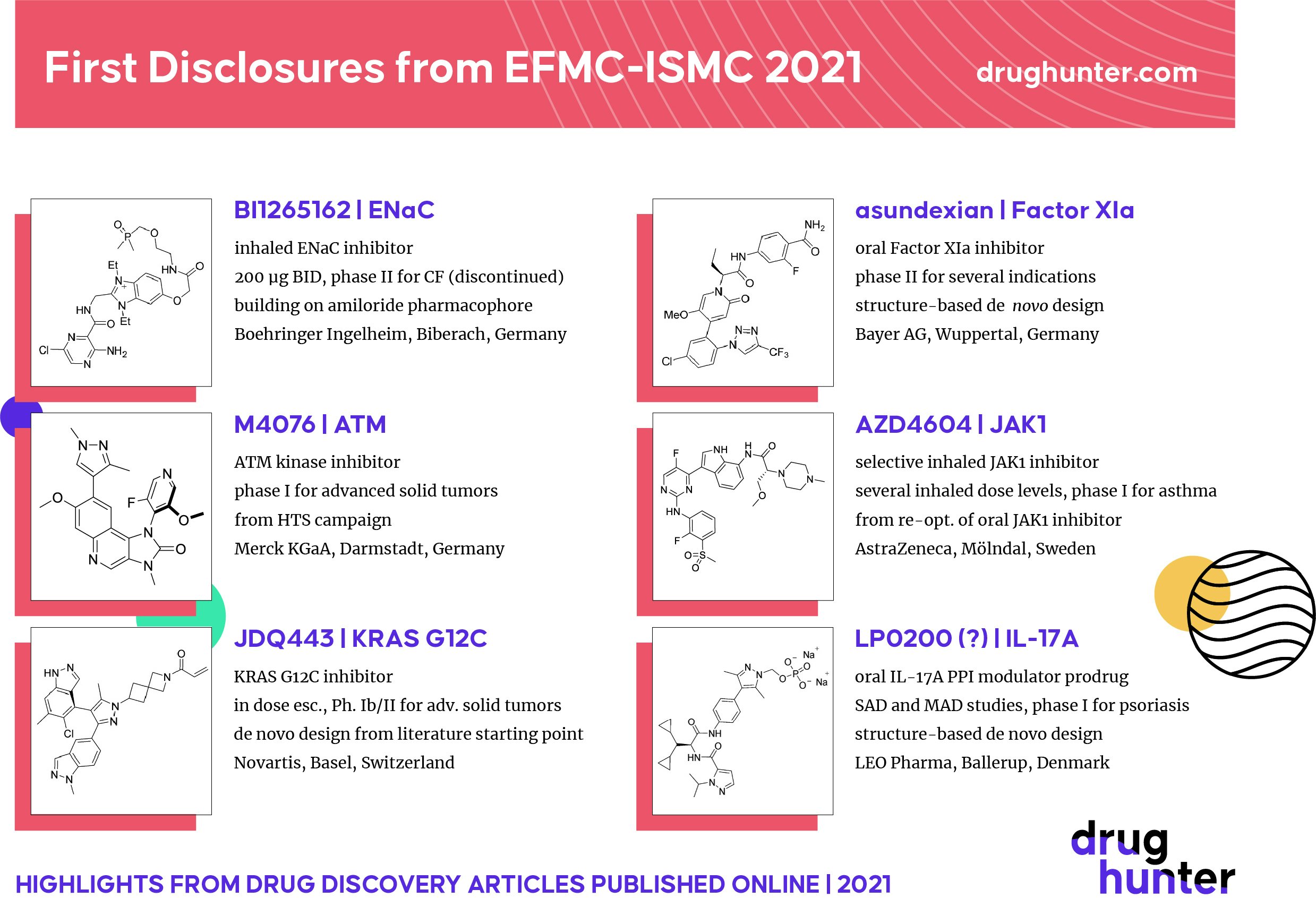
The first disclosures of new drug candidates are always the highlight of medicinal chemistry conferences. Six novel drug candidates were recently disclosed by European companies at the European Federation for Medicinal Chemistry International Symposium on Medicinal Chemistry (EFMC-ISMC) Conference in Basel, including a KRAS G12C inhibitor from Novartis, an inhaled ENAc inhibitor for Cystic Fibrosis from Boehringer Ingelheim, and an IL17A protein-protein interaction modulator from LEO Pharma. Highlights from each talk are summarized below.

First Disclosures of Small Molecule Drug Candidates – EFMC Conference – 2021
BI1265162 – Boehringer Ingelheim Inhaled ENAc Inhibitor
The Boehringer Ingelheim (BI) ENAc inhibitor was an inhaled, Ph. II clinical candidate for cystic fibrosis. In cystic fibrosis (CF), mutations in the CFTR channel along with a hyperactivated Endothelial Sodium Channel (ENaC) leads to hyperconcentrated mucus and impaired mucociliarly clearance. This results in bacterial colonization, inflammation and ultimately respiratory failure. Therefore, ENAc inhibition is a potential mutation-agnostic approach for treatment of CF. Amiloride is a known ENaC inhibitor used for the treatment of high blood pressure but is not suitable for the treatment of cystic fibrosis. The BIteam, represented by presenter Jörg Kley, aimed to develop lung-specific ENaC inhibitors suitable for administration via BI’s Respimat inhaler. Consequently, a low dose, low permeability, very high aqueous solubility and hydrolytic stability were key requirements for the compound. BI1265162, which not only features an unusual positively charged benzimidazolium group but also a phosphine oxide, was eventually selected as a clinical candidate and evaluated as an add-on therapy to standard of care, including CFTR modulators. After demonstrating safety and tolerability in Phase I studies, BI1265162 was advanced into Phase II trials where 200 µg twice daily administration via the Respimat inhaler was evaluated with improvement of FEV1 after 4 weeks of treatment as the primary endpoint. Unfortunately, due to lack of efficacy the compound was discontinued.
Asundexian (BAY 2433334) – Bayer Oral Factor Xia Inhibitor
Susanne Roehrig (Bayer Wuppertal) shared details on the discovery of Asundexian (also known as BAY2433334), Bayer’s factor XIa inhibitor. Following their success with Rivaroxaban (Factor Xa inhibitor), the Bayer team decided to tackle another key target in the coagulation cascade. In contrast to factor Xa, factor XIa is not required for homeostasis. The chemical class was identified using internal X-ray cocrystal structures of two tool compounds with human FXIa. Extensive use of Water Map analysis helped the team identify the Ester Binding Pocket and the P1’ pocket as key areas to investigate SAR for. A sub-micromolar hit was optimized to their first clinical candidate, which had a carboxylic acid. Unfortunately, in a phase I clinical trial in healthy volunteers, the compound showed a very short half-life in humans. To address this, the team decided to replace the carboxylic functional group filling the P2’ pocket with a non-acidic substituent. After several rounds of optimization, BAY2433334 (asundexian) was identified as a second clinical candidate. Asundexian is a highly potent and selective FXIa inhibitor suitable for once daily dosing. Asundexian is currently being tested in more than 4000 patients in several phase 2 studies (NCT04304534, NCT04510987, NCT04304508, NCT04218266).
M4076 – Merck KGaA ATM Kinase Inhibitor
DNA damage repair pathways for cancer treatment have attracted a lot of attention in the past decade since the approval of PARP inhibitor olaparib. Several kinase inhibitors targeting DDR pathways (such as ATR or DNA-PK inhibitors) are currently being clinically investigated in clinical trials. ATM is a protein kinase from the PIKK family that plays an important role in DNA damage repair mechanisms. ATM is mainly involved in regulating the cellular response to DNA double-strand breaks. Thomas Fuchß (Merck KGaA) shared the work from Merck colleagues on their discovery of M4076, a highly potent and selective ATM inhibitor currently in a phase I clinical trial for patients with advanced solid tumors. The initial chemical series was initially identified from a HTS screening campaign on the other DDR target DNA-PK. During the lead optimization phase, the team worked intensively on improving the solubility of their kinase inhibitors. These efforts led to the identification of M4076, a stable atropoisomer, that was suitable for clinical development. M4076 also showed synergistic efficacy in several preclinical models, especially in combination with radiation therapy. (NCT04882917)
AZD4604 – AstraZeneca Inhaled JAK1 Inhibitor
JAK/STAT signaling is involved in several key inflammatory pathways in asthma and has therefore been targeted with small molecules by several companies in the past. Magnus Nilsson from AstraZeneca presented the discovery of AZD4604, a potent and selective Janus Kinase 1 Inhibitor, an inhaled treatment for respiratory disease. The AZ team aimed to discover inhibitors with a high selectivity for JAK1 (in order to avoid erythroid toxicities) and which are suitable for inhalative administration to minimize systemic immune suppression. Building on a number of very potent and selective JAK1 inhibitors from an earlier program, further optimization was done with emphasis on properties like crystallinity, melting point and lipophilicity while maintaining high potency. AZD4604 is a single digit nanomolar inhibitor of JAK1 and was found to be >100 fold selective over a panel of 351 kinases. AZD4604 shows favorable lung PK as well as target engagement (inhibition of STAT3/5 phosphorylation) and will be tested in a Phase 1 clinical trial (NCT04769869).
NVP-JDQ443 – Novartis KRAS G12C Inhibitor
While KRAS is considered one of the most important oncogenes, it was considered undruggable for many decades. After a landmark paper from the Shokat lab (UCSF) in 2013, the race to identify covalent KRAS G12C inhibitors started. Several companies tried to tackle this challenge and Amgen was successful in identifying sotorasib, the first, and currently only, KRAS G12C inhibitor approved by the FDA (in May 2021). Simona Cotesta from Novartis presented the work on the identification of NVP-JDQ443, a covalent KRAS G12C inhibitor. Like other KRAS G12C inhibitors, NVP-JDQ443 traps the GDP-bound inactive conformation of KRAS. The pyrazole-containing hit compound class was identified using de novo design starting from in-house crystal structures. Further exploration of pyrazole substituents and replacement of the central phenyl linker by a spiro-piperidine resulted in both increased cellular activity and lower chemical reactivity of the acrylamide warhead. An X-ray cocrystal structure also highlighted a novel binding mode for the molecule under the Switch II pocket of KRAS. NVP-JDQ443 is a potent and mutant-selective KRAS G12C inhibitor that is currently being investigated in a phase Ib/II clinical trial of JDQ443 alone, in combination with TNO155 (SHP2 inhibitor) and in combination with PD-1 blockade in patients with advanced solid tumors harboring the KRAS G12C mutation (NCT04699188)
LEO Pharma Oral IL-17 Modulator
Il-17 signaling plays a major role in the pathogenesis of Psoriasis vulgaris, a common chronic inflammatory skin disease. Both Anti-IL-17A and anti-IL-17RA monoclonal antibodies (secukinumab, ixekizumab and brodalumab) have been approved as highly effective treatments for psoriasis. The LEO Pharma team, represented by Kevin Dack, aimed to develop an orally bioavailable, small molecule IL-17A protein-protein interaction modulator (PPIm) with comparable efficacy & safety to the available mAbs. A series of macrolide inspired macrocycles was initially investigated but was eventually stopped. LEO scientists then established another series (featuring a benzhydrylglycine core) through structure-based design, inspired by overlaying lead compounds from LEO and Pfizer. Most notably, replacing both phenyls in their earlier lead with bis-cyclopropyl not only decreased MA and logP but also led to a gain in potency and favorable ADME. Final optimization of the terminal amide led to their IL-17A PPIm which was further elaborated into a phosphate prodrug in order to improve solubility and enable oral absorption. In a mouse imiquimod model, their compound showed dose-dependent inhibition with their small molecule comparable to s.c. mouse IL-17A mAb. One of their oral IL-17 PPIs (the definite structure of the final clinical candidate was not communicated) has been progressed to Phase 1 (NCT04883333).
Hope this was helpful – explore drughunter.com for more drug discovery highlights.