Letters from Drug Hunters #1 – Apr. 2022
One of the privileges that have come with building Drug Hunter has been all the insightful thoughts and commentary our readers share. We also appreciate how readers help catch mistakes and offer corrections. This new series of “Drug Hunter Letters” highlights recent insightful communications from drug hunters, which includes this time:
Dr. Nick Carruthers on the History of PDE4 Inhibitors
Dr. Ingo Hartung on the BRD9 Degrader, CFT8634
Dr. Dennis Koester on the Mechanism of Action and Metabolism of Viloxazine
Johnny Phan with Questions to the Audience On the Relevance of Stereochemistry in IMID-based Degraders
Dr. Nick Carruthers on the History of PDE4 Inhibitors
In our article covering ACS First Disclosures, we mistakenly referred to the Schering AG compound, rolipram, as a Schering-Plough compound. This was caught thanks to Dr. Nick Carruthers, consultant and previously Head of Strategic and Scientific Outreach at Johnson & Johnson. Rolipram, the prototypical PDE4 inhibitor, was patented by Schering AG in the 1970's.
Nick adds, "Rolipram is quite interesting as more recent PDE4 molecules were counterscreened against the 'Rolipram binding site' since this was thought to be the cause of the GI side effects and emesis with the early PDE4s in the 90's. SmithKline Beecham (SB, now GSK) did get a clinical candidate, Ariflo, but I don't believe it ever reached the market.
These papers by Torphy and Duplantier cover the rolipram binding issue, and this Hirose article looks at emesis in a shrew model. Here is a recent review by Li and colleagues, and here is a very recent review in J. Med. Chem. on targeting PDE4. Overall these papers show you how the field evolved and presumably led to a greater understanding of the GI and emetic side effects of the early compounds."

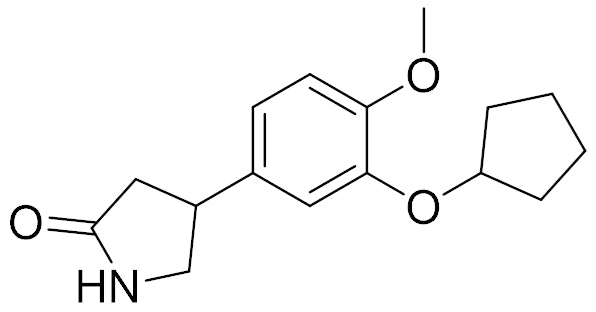
rolipram
Dr. Ingo Hartung on the BRD9 Degrader, CFT8634
In our article covering New Drug Candidates at AACR, we initially omitted the stereochemistry from CFT8634. This was updated thanks to Dr. Ingo Hartung (@HartungIngo) Sr. Director and Head of Medicinal Chemistry GER at Merck KGaA, Germany.
Ingo adds, “CFT8634 is clearly the result of a very systematic optimization campaign covering target binder, E3 binder and the linker. Of note are the modified CRBN binder (aniline linkage), the redesigned BRD9 binder compared to initially described tool degraders and a relatively short and rigidified linker. Overall, these changes reduced molecular weight, number of H bond donors, rotatable bonds, number of aryl rings leading to a highly potent degrader with oral exposure across species and selectivity against BRD4.
P.S. The lecture was excellent. In case you are looking for speakers for case studies, you should put her [Dr. Katrina L. Jackson of C4 Therapeutics] on the list."


CFT8634
Dr. Dennis Koester on the Mechanism of Action and Metabolism of Viloxazine
Recently, we published our “Deep Dive” on 2021’s Neurology Drug Approvals. Dr. Dennis Koester, a Principal Scientist II of Novartis, Emeryville, writes:
“It is remarkable that more than a decade has passed since a new medication was approved for the incredibly common disease, ADHD, and that this changed with a molecule (viloxazine) that was discovered in the 1970’s. It had been approved for depression in a few European countries, and numerous studies have since supported viloxazine’s repurposing for ADHD.
One of the many fascinating aspects of this ‘new-old’ medication is the complex mechanism of action synergistically driving up the serotonin (5-hydroxytryptamine, 5-HT) concentrations in the pre-frontal cortex (PFC). Viloxazine shows an IC50 of 0.26 μM on the norepinephrine transporter (NET). Serotonin uptake was evaluated and resulted in negligible activity with an IC50 of 257 μM. A screen of relevant serotonin receptors found viloxazine to be a 5-HT2C agonist with an EC50 32.0 μM, and a 5-HT2B antagonist with an IC50 of 27.0 μM.
Although these numbers seem high, at clinically relevant doses of 400 mg, >93% receptor occupancy is predicted for NET, 5-HT2B,and 5-HT2C. In an in vivo rat brain microdialysis model, which looked at extracellular concentrations of several neurotransmitters including NE, dopamine, and serotonin, an increase of peak concentrations in the PFC by 500-670% over baseline was observed for all three neurotransmitters at a dose of 50 mg/kg. Viloxazine’s combined effect on these neurotransmitters likely contributes to its pharmacological effect and efficacy in the clinic.
Another very interesting aspect of viloxazine is its metabolism. Viloxazine is mainly metabolized by CYP2D6 to 5-hydroxyviloxazine (5-HVLX) and is subsequently glucoronidated to 5-HVLX-gluc mediated by uridine 5'-diphospho-glucuronosyltransferases. Interestingly, Viloxazine exposure could be increased by about 35% when co-dosed with CYP2D6 inhibitor paroxetine. In genetically poor CYP2D6 metabolizers exposures increased up to 25%. Viloxazine is rapidly metabolized and excreted in urine with no known active metabolites.”

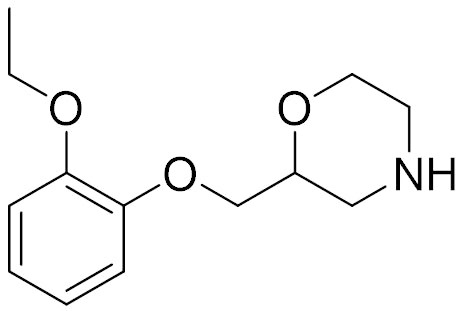
viloxazine
Questions to the Audience: On the Relevance of Stereochemistry in IMID-based Degraders
Johnny Phan, Biotech Associate and previously at Novartis (NIBR) and Eli Lilly, asks:
"Shouldn't the chiral center (on the imide) on CFT7455 readily epimerize?" Why would a company develop a single isomer if this is the case?


CFT7455
“Other degraders such as CC-92480 appear to racemize more quickly in vitro than in vivo. Does anybody know the mechanism of the in vivo epimerization?”

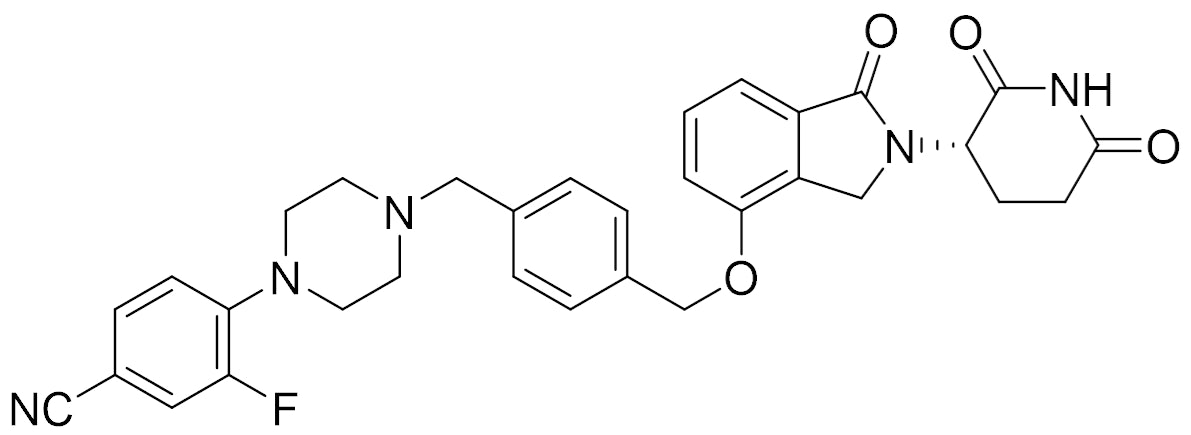
CC-92480
We hope you enjoyed reading as much as we did! Have an answer or something else to share?