In Context: First-Time Disclosures of ACS Spring 2022
The ACS first disclosures from ACS Spring 2022 MEDI section included six molecules, summarized below. Together with industry veteran Jesse Keicher, we put these molecules in context, exploring what makes each molecule interesting to drug hunters beyond what was shared at the conference.
The first disclosures were:
AMG 650, Amgen’s kinesin family member 18A (KIF18A) inhibitor
KVD900, KalVista’s plasma kallikrein inhibitor
LY3372689, Lilly’s OGA inhibitor
PF-07038124, Pfizer’s topical PDE4 inhibitor
AZD9574, AstraZeneca’s PARP1 inhibitor
BLU-945, Blueprint’s EGFR triple-mutant inhibitor

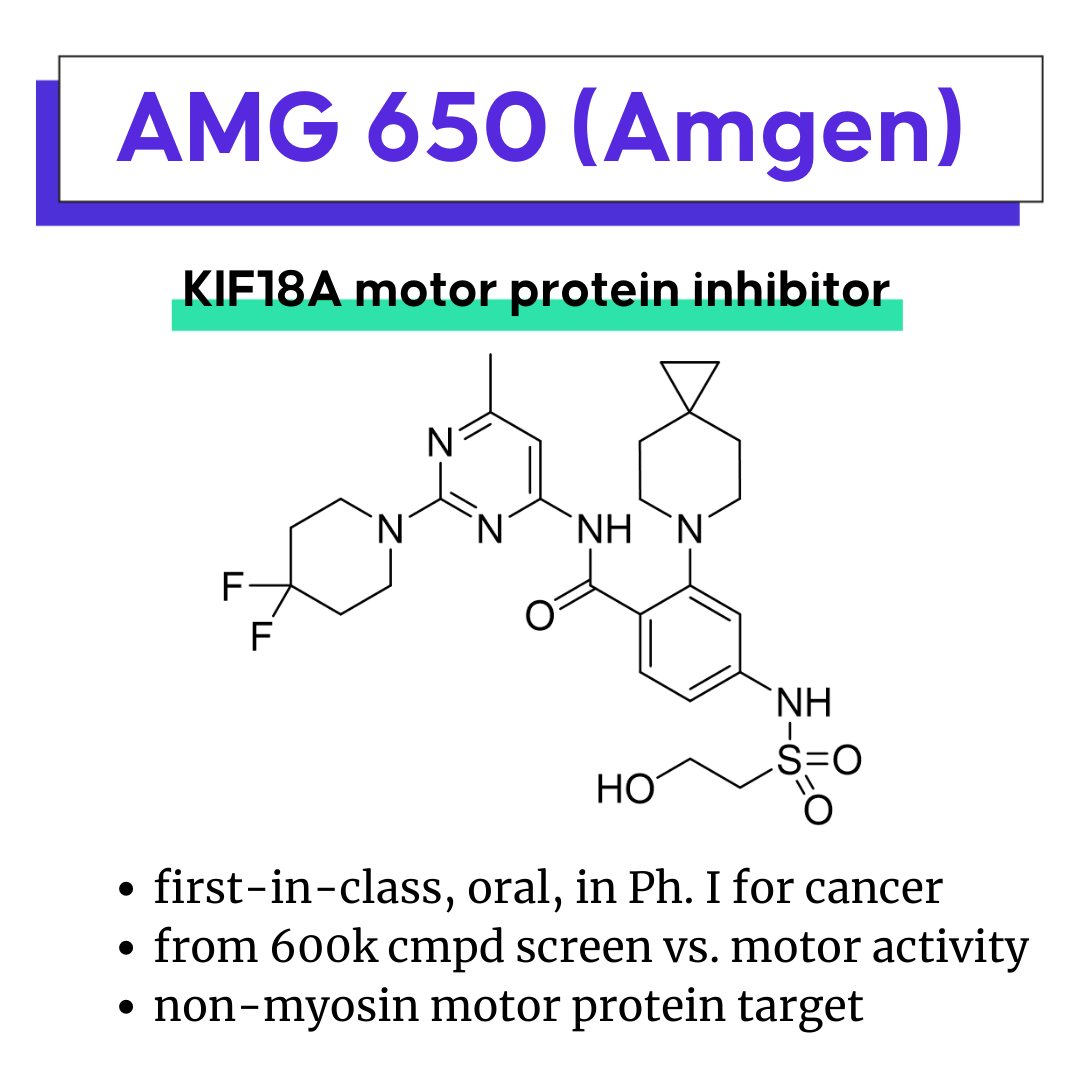
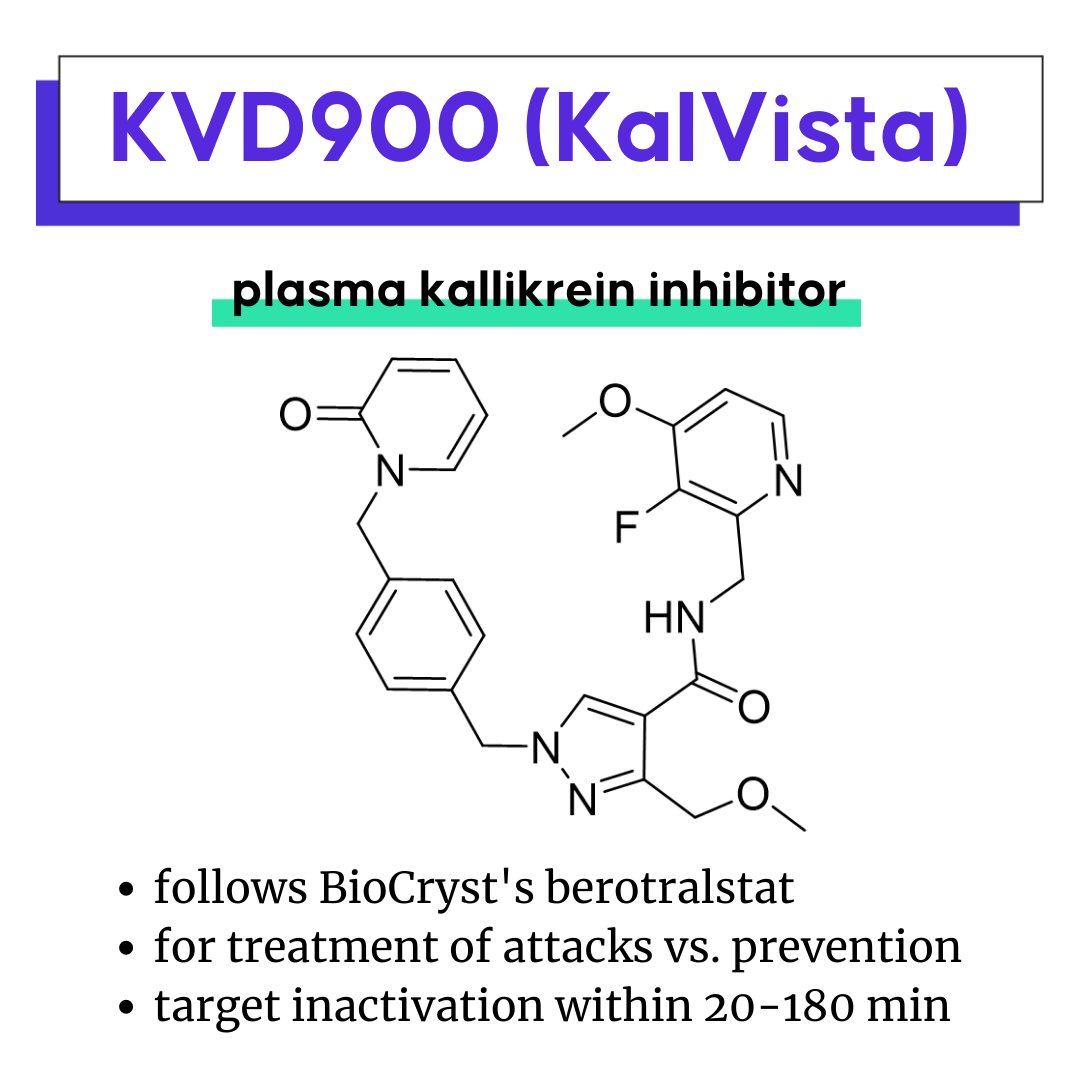
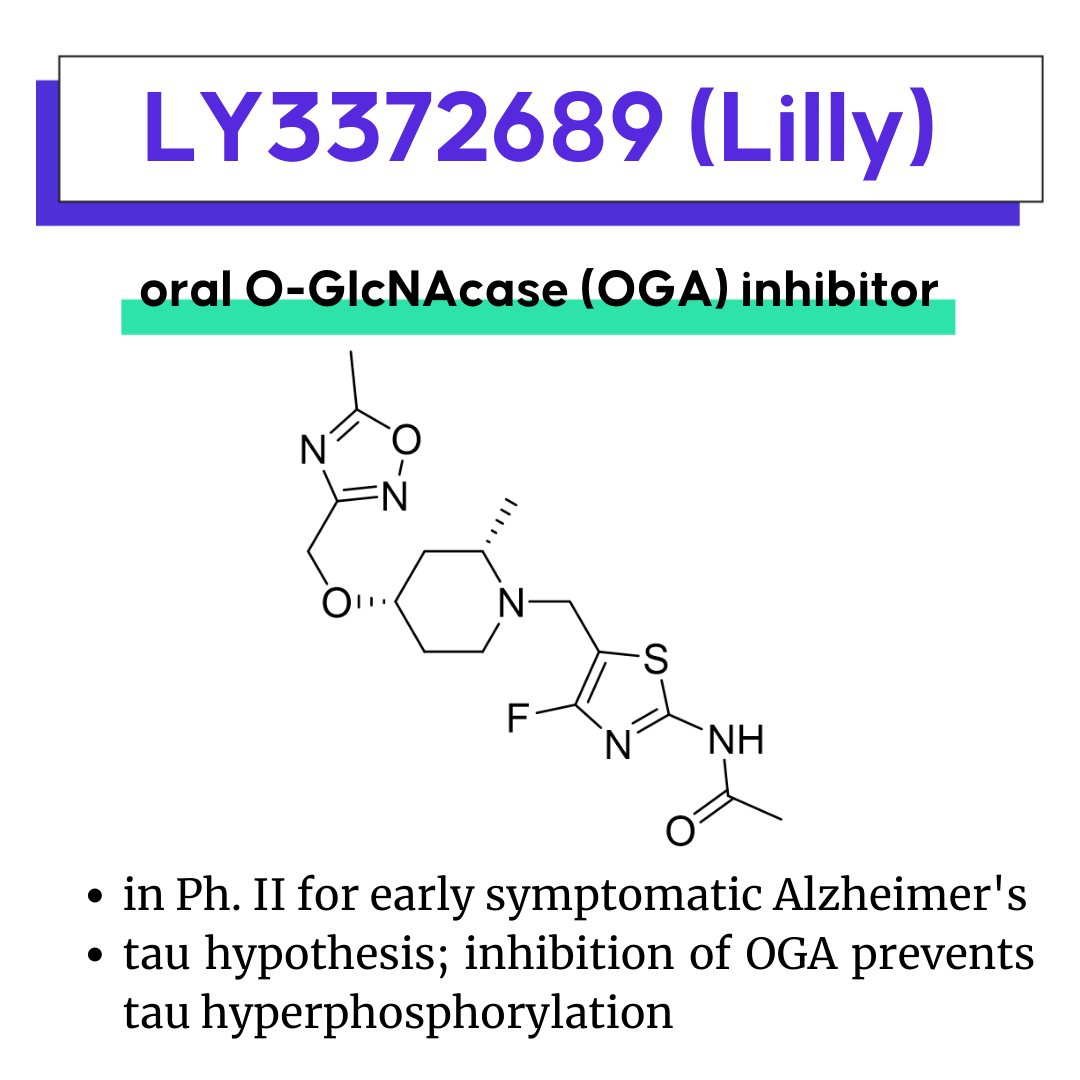
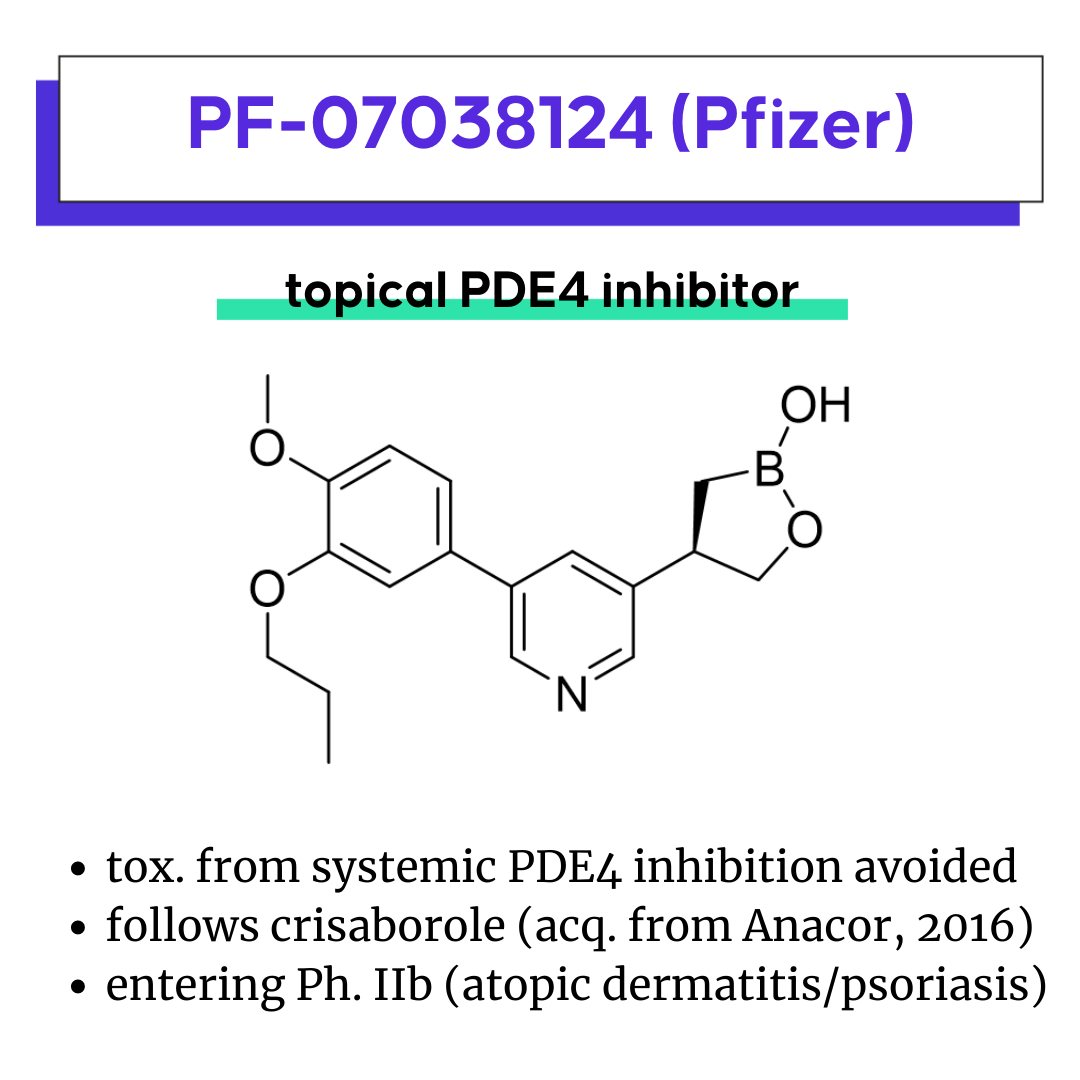
AMG 650: A first-in-class motor protein inhibitor for cancer

AMG 650 is Amgen’s oral, first-in-class, selective inhibitor of kinesin-like protein 18A (KIF18A) in Ph. I in cancer patients to identify the MTD or RP2D. Amgen recently published related efforts on this novel target.
KIF18A is a mitotic kinesin motor protein that regulates chromosome positioning during cell division and is overexpressed in a subset of human cancers. Motor proteins are a relatively novel target class that have recently been proven to be clinically actionable by myosin inhibitors such as Myokardia’s mavacameten and activators such as omecamtiv mercabil (Cytokinetics).
Amgen’s systematic strategy to tackle this target and advancement of a KIF18A inhibitor into the clinic outside of cardiology shows motor proteins like myosin are not one-offs in target class, but the class may be more broadly amenable to the industrial drug discovery process. We may start to see more startups and pipeline programs on motor protein drugs in general.
In preclinical studies, KIF18A gene inactivation suggests that its function can be dispensable for normal somatic cell division making KIF18A an interesting novel therapeutic target for treating chromosomally unstable cancers while sparing non-transformed cells.
KVD900: A fast-acting, follow-on plasma kallikrein inhibitor
KVD900 is potent and selective orally-available plasma kallikrein (PK) inhibitor for the treatment of angioedema attacks in hereditary angioedema (HAE) (Ph. III by KalVista Pharmaceuticals). Another kallikrein inhibitor, berotralstat, was recently approved for HAE attack prevention and featured as a 2021 Molecule of the Year candidate. Kallikrein inhibitors prevent the breakdown of high molecular weight kininogen (HK) and subsequent buildup of bradykinin, an underlying cause of the disease.
A differentiation angle for KVD900 is its development as an on-demand treatment for angioedema attacks rather than prevention, with a PK and property profile that supports rapid inactivation of the target between 20 min and 3 h. The recently approved oral kallikrein inhibitor, berotralstat, is specifically limited in the label against use in acute HAE attacks.
To demonstrate this fast-acting target product profile, the company developed a capillary-based immunoassay that provides a rapid (<3 hrs) and semi-automated method to quantify HK in human plasma 5-fold faster than traditional western blotting. This method was used to assess its rapid pharmacodynamic effects in clinical trials. Measurements of plasma HK have largely relied on traditional western blot immuno-assays, which are labor intensive and associated with high inter-assay signal to background variability.
In the Ph. II trial, KVD900 met its primary endpoint of reduced rescue within 12 h; in the Ph. III trial, the different endpoint of reduced time to beginning of symptom relief is being used (also met in Ph. II). Ph. III data is expected at the end of 2023, with a potential NDA filed in 1H 2024.
LY3372689: Another test of the tau-hypothesis
LY3372689 is an oral O-GlcNAcase (OGA) enzyme inhibitor in development by Eli Lilly and Company. LY3372689 is currently in phase II to assess the safety, tolerability and effect in participants with early symptomatic Alzheimer's Disease (AD). Alzheimer's Disease is hypothesized to be a tauopathy, characterized by the accumulation of insoluble, hyperphosphorylated tau aggregates, the main component of neurofibrillary tangles (NFTs).
O-GlcNAcylation or the addition of N-acetylglucosamine is a post-translational protein modification that occurs on serine and threonine residues. Since these are also the sites of hyperphosphorylation, O-GlcNAcylation competes for the same sites. Inhibition of O-GlcNAcase (OGA), the enzyme responsible for the removal of O-GlcNAc, has been shown to increase O-GlcNAcylation of tau and thereby prevent hyperphosphorylation stabilizing tau in a soluble, nonpathogenic form.
LY3372689 is an example of the shift in Alzheimer’s drug discovery focus from testing the Aꞵ hypothesis to tau and other hypotheses, due to the large numbers of clinical failures targeting the beta-amyloid pathway (e.g. BACE1, gamma-secretase, and Aꞵ antibodies depending on who you ask). Tau is currently believed to be more closely linked to cognitive degeneration. LY3372689 is among the second generation of oral small molecule drugs that target the tau pathway following failures of first-generation tau-targeted antibodies.
PF-07038124: Avoiding mechanism-based toxicity by tissue-restriction
PF-07038124 is a PDE4 inhibitor for topical administration in development by Pfizer. It recently completed a phase IIa study to assess efficacy, safety, tolerability and pharmacokinetics of PF-07038124 ointment in participants with atopic dermatitis or plaque psoriasis in EMPORIA study.
Systemic PDE4 inhibition is associated with mechanism-based GI tox., first identified in the 90’s with Schering AG’s rolipram. Previously approved PDE4 inhibitors include ibudilast (oral, broad PDE inhibitor used in Japan for 20+ years), roflumilast (oral, 2010 in EU for severe COPD), apremilast (oral, 2014 for psoriatic arthritis and plaque psoriasis), and crisaborole (topical, 2014 for treatment for psoriasis and atopic dermatitis). Hence, strategies to manage the narrow therapeutic window with approved PDE4 inhibitors have been to focus on severe disease, employ dose titrations, or most recently for crisaborole, use a tissue-restricted drug. Crisaborole was acquired by Pfizer through Anacor in 2016.
With a dermatology landscape that is evolving to have new antibodies like anti-IL-4Rɑ dupilumab and anti-IL-13 tralokinumab as strong options, combinability of a PDE4 inhibitor will be important. With the crisaborole patent expiring in a few years, there’s room for a next generation PDE4 that has time to be tested in different combinations and settings. Though there is not enough published to differentiate PF-07038124 and crisaborole, Pfizer does have an ongoing trial comparing the two in vitiligo with phototherapy, where a much lower topical concentration (0.01% vs. 2%) of PF-07038124 is needed.
Utilizing a “topical by intent” approach, Pfizer focused on high target potency, high systemic clearance, and suitable solubility and permeability parameters acceptable for topical formulation. This led to the discovery of PF-07038124 as a novel chiral oxaborole-based PDE inhibitor. The novelty and synthetic complexity of these chiral oxaboroles required invention of new synthetic approaches since there were no reported asymmetric routes to the oxaborole ring system contained within PF-07938124.
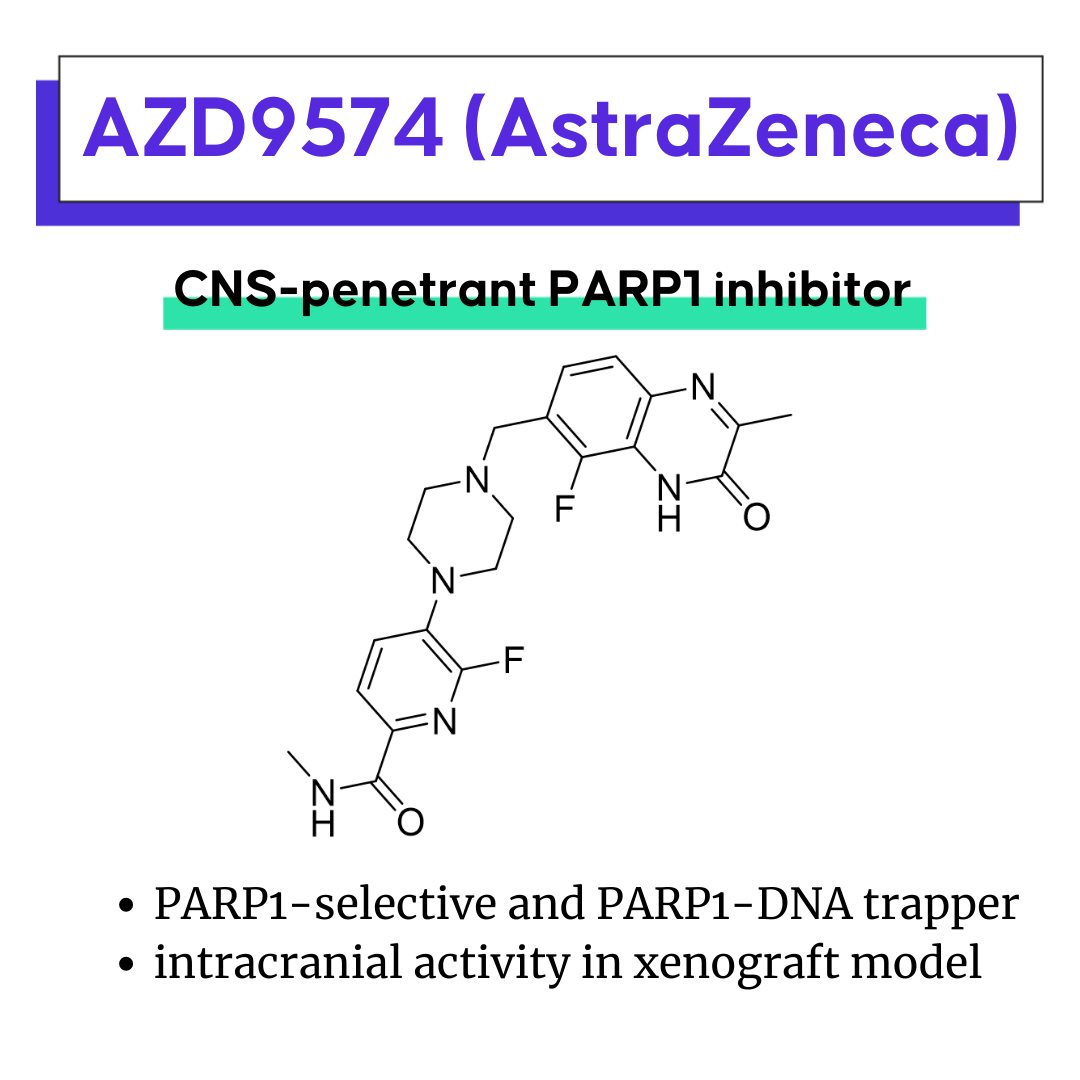
AZD9574: A next generation PARP inhibitor for brain metastases
AZD9574 is a preclinical, novel, second-generation CNS-penetrant PARP1-selective inhibitor and PARP1-DNA trapper from AstraZeneca. PARP inhibitors kill cancer cells by targeting tumor cell DNA damage repair mechanisms which are redundant in healthy cells (synthetic lethality). However, most approved PARP inhibitors cannot cross the blood-brain barrier, limiting their application in treating brain tumors and metastases.
AZD9574 was designed to cross the blood brain barrier to enable the targeting of primary and secondary brain malignancies. Structure-based and property-based techniques were used to identify potent leads from an in-house database which were optimized into CNS penetrant second generation PARP1 selective inhibitors and trappers.
In early preclinical studies, AZD9574 demonstrated activity in an orthotopic, intracranial xenograft model with aberrant DNA repair.
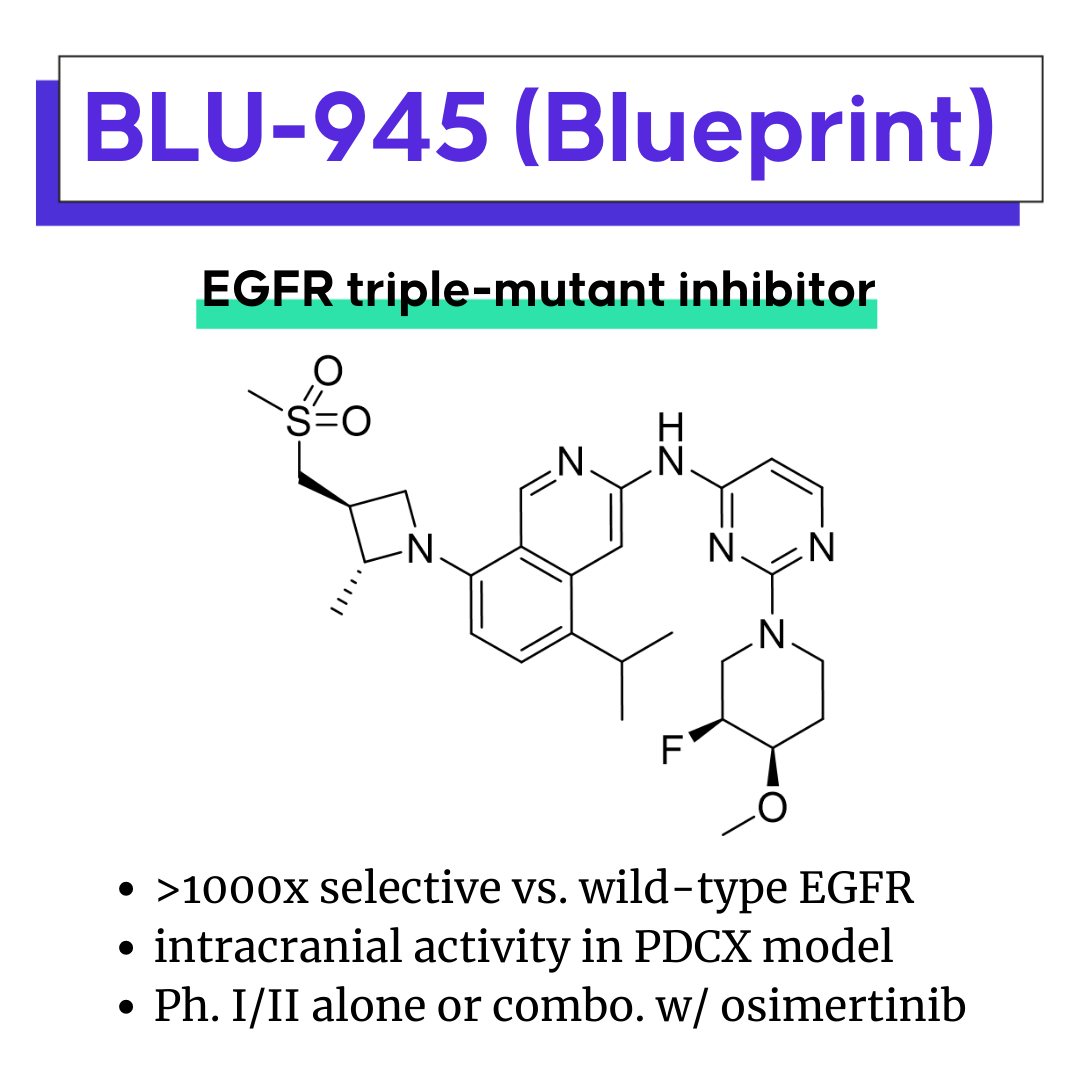
BLU-945: A potential best-in-class mutant EGFR inhibitor
Nearly two decades after the approval of EGFR inhibitor gefitinib in 2003, mutants of EGFR remain important cancer targets. BLU-945 is an oral, selective and potent fourth generation (4G) EGFR inhibitor of double-mutant or triple-mutant EGFR (T790M or ex19del/T790M/C797S) in clinical development by Blueprint Medicines. Currently it is in Ph. I/II to evaluate safety, tolerability, and PK/PD as monotherapy or in combination with third generation (3G) EGFR inhibitor, osimertinib. In an enzyme assay, it is >1000x selective against WT EGFR, and appears to be brain-penetrant, having demonstrated intracranial activity in a patient-derived cell xenograft model.
T790M and C797S mutations are the most common on-target resistance mechanisms to first generation (1G) EGFR inhibitors and third generation (3G) EGFR inhibitors, respectively. Currently, there are no approved therapies for patients with disease progression following treatment with 3G EGFR inhibitors.
We hope you found this summary helpful. If you attended ACS or another recent conference, what was the most interesting you saw? Let us know by submitting a Letter to the Editor!