Drug Hunter Letters #3 - September 2022
This edition of Drug Hunter Letters highlights recent insightful communications from Drug Hunter readers, including:
Simon Bury on the History of GSK’s Recently Acquired $1.9B JAK Inhibitor
Callie Bryan and Bryan McKibben on Insulin Receptor Partial Agonists and Recent Oncology Highlights
Jesse Keicher on CXCR1/2 Antagonists in Treating Neuropathic Pain
Correction from Abbas Kazimi: On the Binding Mode of NDI-034858
Leon Wang Asks: Why Would the FDA Rescind a Breakthrough Designation?
Simon Bury on the History of GSK’s Recently Acquired $1.9B JAK Inhibitor

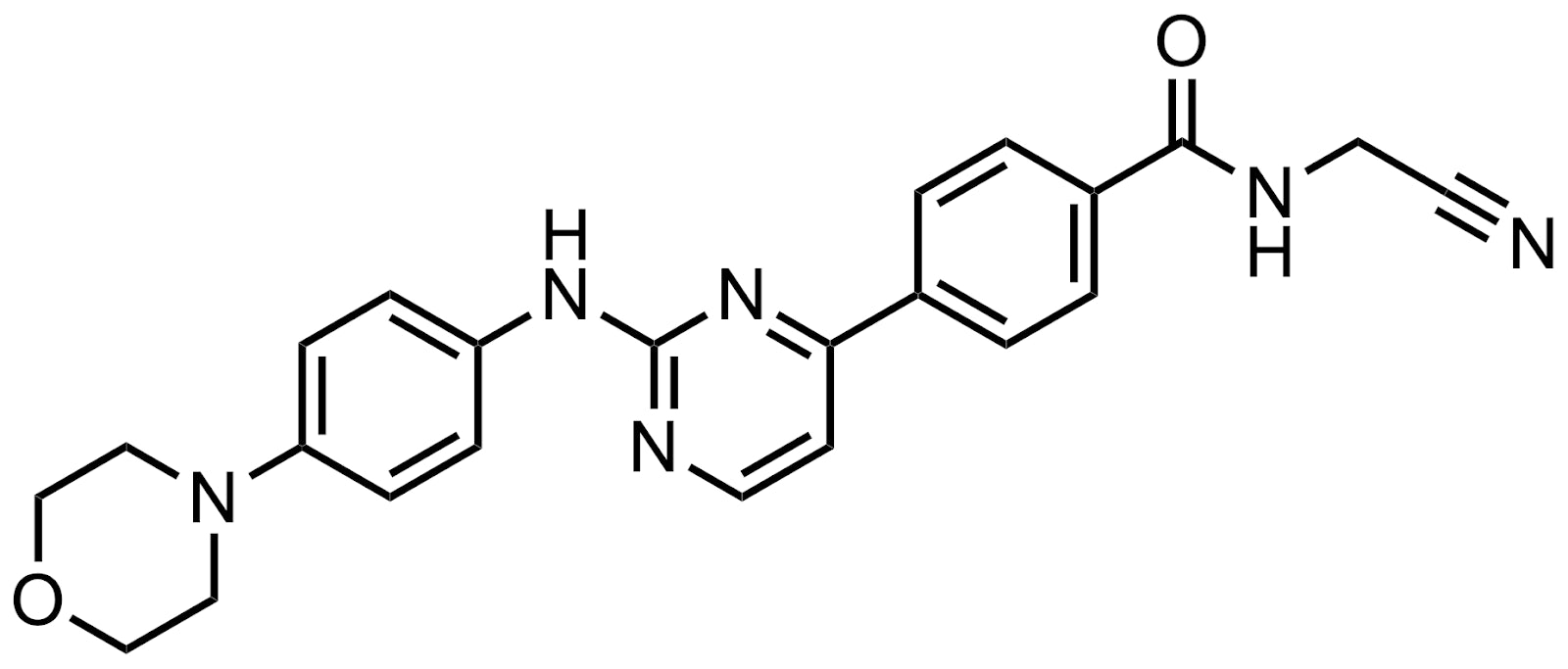
Momelotinib (CYT387)
We recently published an article about why GSK acquired a JAK inhibitor for $1.9B from Cytopia. Simon Bury, Global Head of Business Development of SYNthesis Med Chem, shares some more history:
“SYNthesis med chem was established by a number of the scientists that previously worked for Cytopia in Melbourne and discovered momelotinib. These included Professor Andrew Wilks, who was the co-founder and CSO of Cytopia. Andrew was previously an academic scientist at the Ludwig Institute for Cancer Research in Melbourne, where he discovered JAK1 and JAK2, and it was Andrew that initially came up with the name JAK as they were “just another kinase.”
“After Cytopia wound down their research activities in 2007, Andrew and Dr. Xian Bu (a Senior Scientist at Cytopia) co-founded SYNthesis med chem as a discovery chemistry-focussed contract research organization with operations in Melbourne and Shanghai. SYNthesis med chem has grown over the years and is now a global chemistry CRO providing custom synthesis, medicinal chemistry & integrated drug discovery services and has operations in China (Suzhou, Shanghai & Chengdu), Australia, North America (USA & Canada), and the UK. The company currently employs over 350 chemists and was acquired by Viva Biotech in 2021.”
We’ll be hosting Andrew Wilks and his Cytopia co-founder Chris Burns on a Drug Hunter Flashtalk later this year - WATCH NOW
sign up for the series, so you don’t miss it!
Callie Bryan on Insulin Receptor Partial Agonists and Recent Oncology Highlights

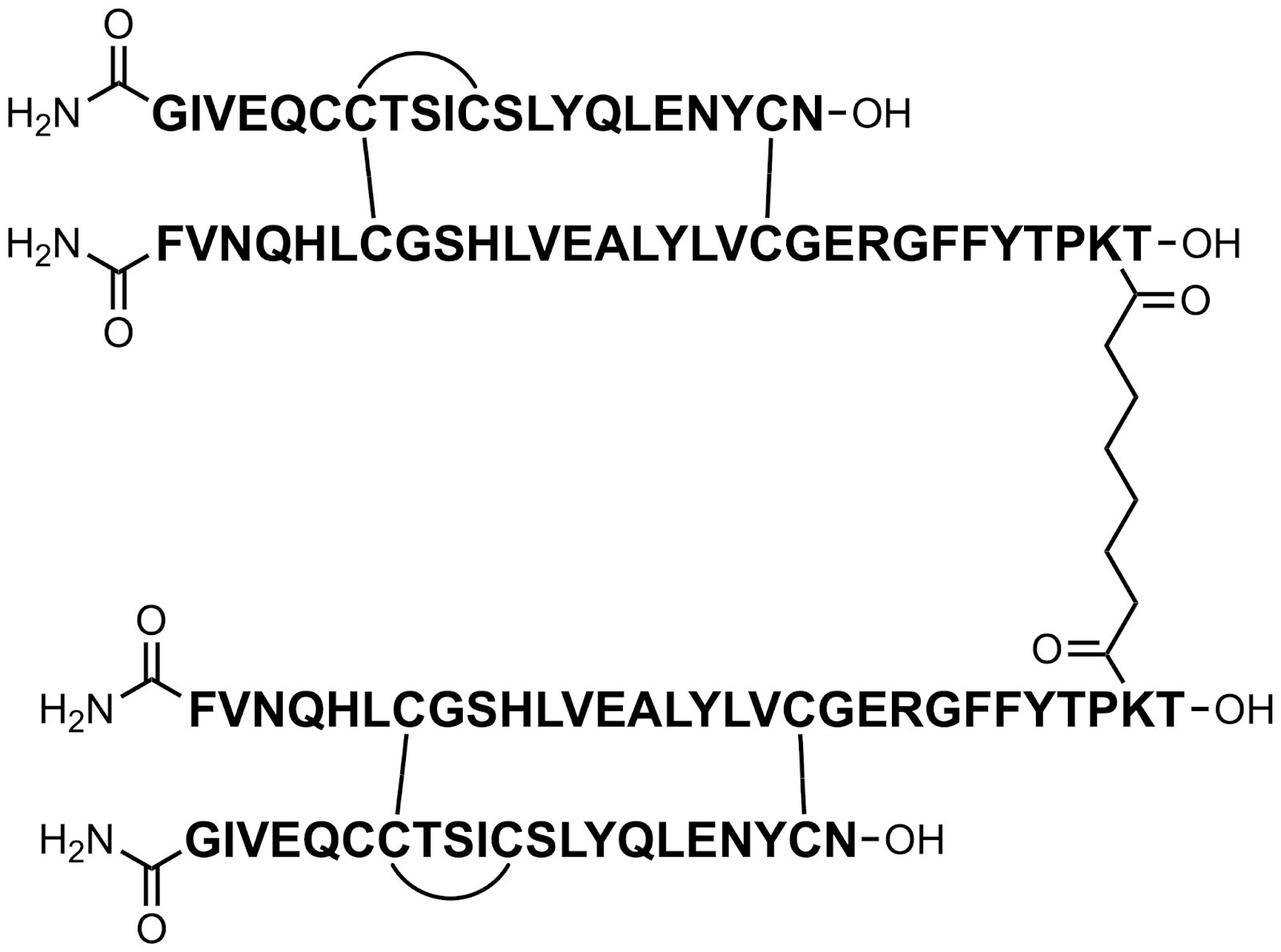
insulin receptor (IR) partial agonists
Two reviewers, Callie Byran and Bryan McKibben found Merck’s Insulin Receptor Partial Agonist Peptides scientifically interesting. Callie said,
“For the insulin receptor (IR) partial agonists, safe and effective treatment of Type 1 diabetes remains a significant unmet medical need. Since its discovery, IR has always been about threading the needle between efficacy and hypoglycemia (safety). Merck’s approach, targeting a partial agonist profile by linking two native insulin receptors with variable tethers, led to 20~70% compared to native insulin. Further development gave two molecules with structurally distinct linkers and capping, MK-1092 and MK-5160. These showed both efficacy in advanced species models and, due to their lower agonism levels (30~40%), attained a wider safety margin over hypoglycemia. It remains to be seen if this will translate to the clinic, but it is definitely promising.”

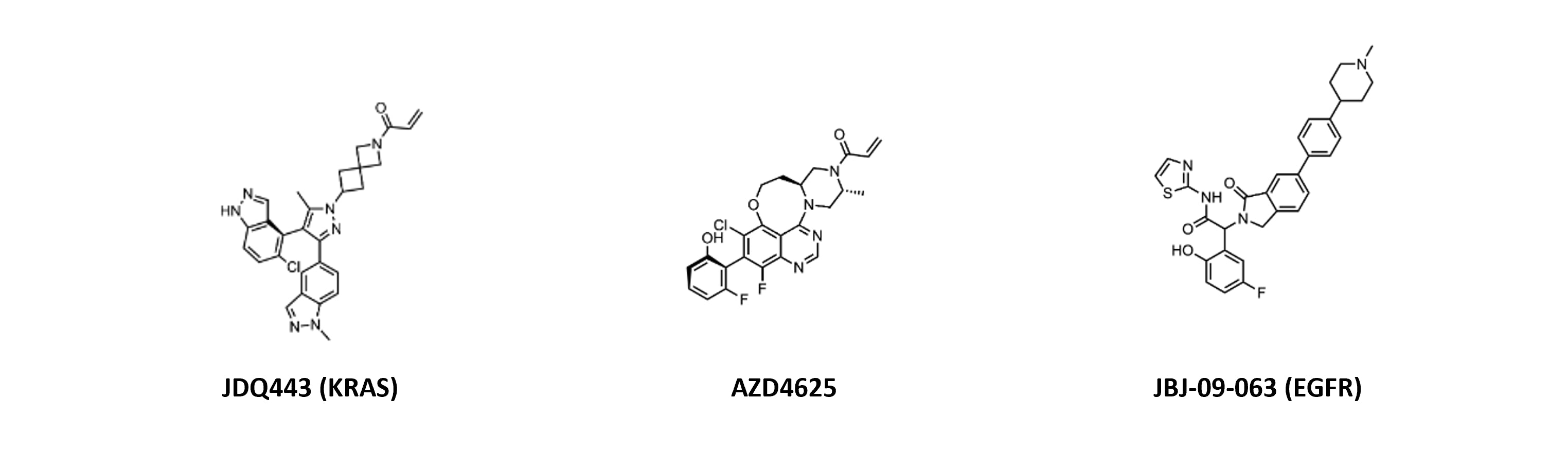
In April’s Molecules of the Month slide deck, Callie also found the KRAS molecules and allosteric EGFR inhibitor interesting:
“Biggest to me are the Novartis and AZ KRAS publications: both use the standard covalent paradigm with small molecules in clinical development but differ significantly in structure and binding. Novartis’ JDQ443 features two indazoles, one in the switch II pocket and the other almost planar to the pyrazole core, occupying different regions of the pocket and exploiting new interactions. This leads to potent binding of the GDP-bound KRAS G12C. AZD4625 also potently inhibits G12C, but through a quinazoline tethering strategy to lock out a bio-relevant binding conformation and researchers addressed extrahepatic clearance.
Regarding [the EGFR inhibitor], JBJ-09-063 provides a very subtle change (N-methyl piperidine) over the previously reported JBJ-04-125-02 (piperazine). Despite similar binding modes, JBJ-040125-02 was significantly more potent with improved oral bioavailability. This gives comparable in vivo efficacy to osimertinib in a patient-derived DFCI52 xenograft model and superior results in C797S models. Further development is still required for clinical success of an allosteric EGFR inhibitor, though.”
Jesse Keicher on CXCR1/2 Antagonists in Treating Neuropathic Pain

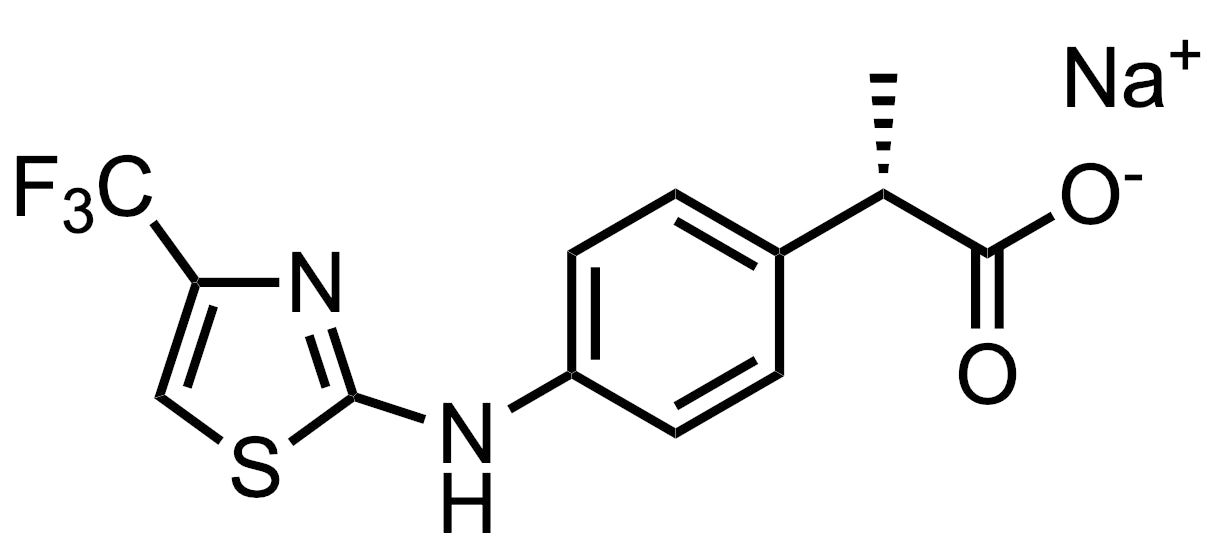
DF2755A
One molecule that was nominated for Molecules of the Month but not selected is DF2755A. Jesse Keicher, however, found this molecule interesting:
“I think DF2755A is worthy of a mention due to the novel use of CXCR1/2 antagonists in treating neuropathic pain, including the possibility of use for chemotherapy-induced peripheral neuropathy. There is a huge unmet need for non-opioid analgesics, and this is the first time a CXCR1/2 antagonist is in clinical trials for pain.
It's not super important, but definitely caught my attention because I had not previously seen a chemokine receptor antagonist for non-inflammatory neuropathic pain. It's not too far of a stretch to think about inhibiting neutrophil cellular influx (via blocking chemokine receptors) to alleviate pain due to inflammation, but what they claim here is that chemokine inhibition can be an effective analgesic in non-inflammation-based pain....that was interesting and new info to me.”
The molecule was not selected due to reviewers’ uncertainty about whether the effects are sufficiently strong relative to other mechanisms and whether the observed effects are on-target.
Correction: Binding Mode of Nimbus TYK2 Inhibitor NDI-034858
In our recent coverage of the First Disclosures from ACS Chicago 2022 we initially incorrectly referred to the Nimbus molecule as a "Type I" binder, it is technically a Type IV kinase inhibitor as it binds to the allosteric, inactive JH2 domain, though its binding mode to the pseudokinase domain is similar to Type I molecules (hinge + away from gatekeeper).
Thanks Abbas Kazimi from Nimbus for alerting us!
Leon Wang Asks: Why Would the FDA Rescind a Breakthrough Designation?

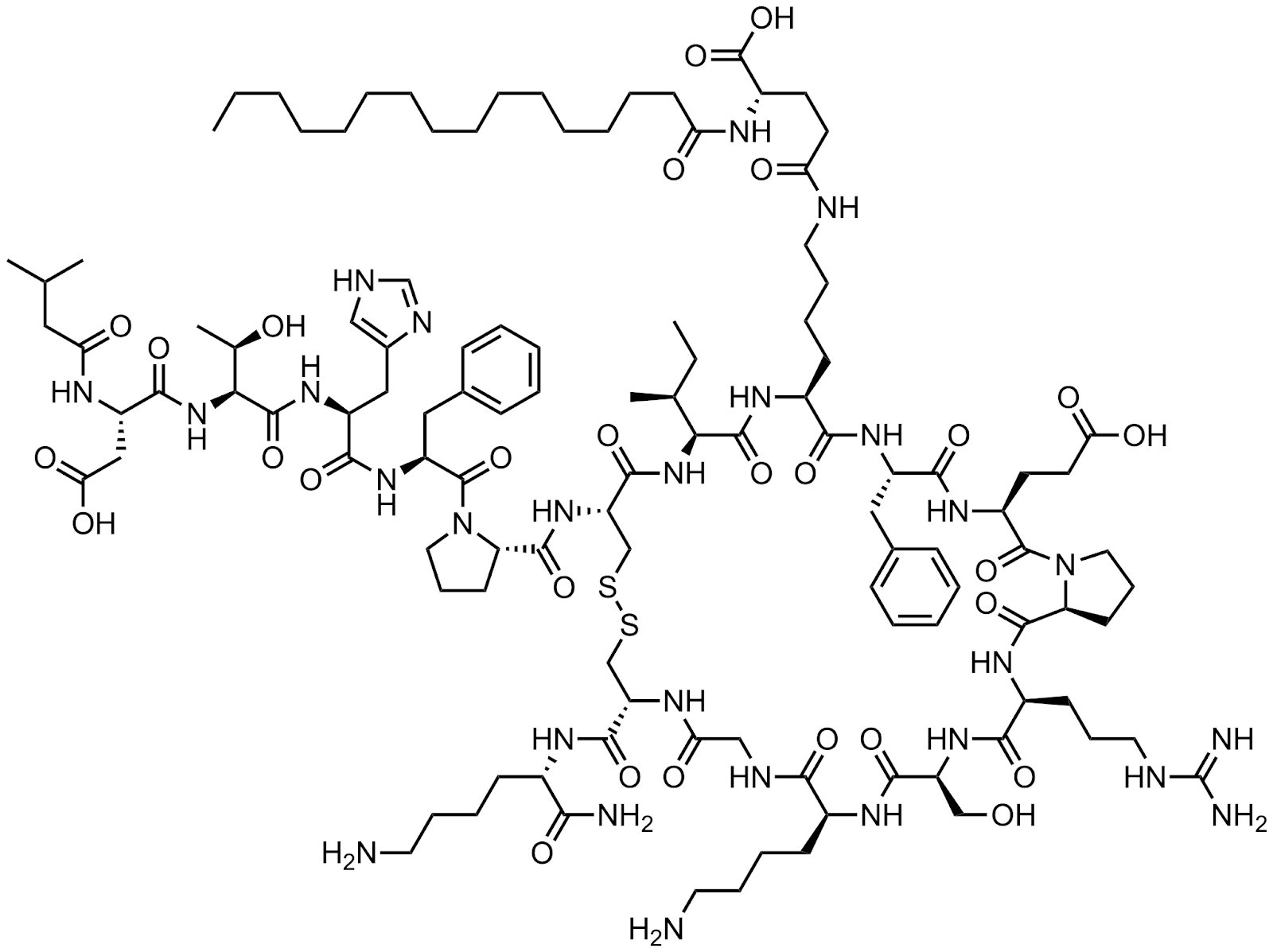
rusfertide
In April, Protagonist Therapeutics received a letter from the FDA about rescinding their Breakthrough Designation (BTD) for rusfertide in polycythemia vera.
Leon then asked, “Why would the FDA give a BTD and then rescind it later? Does this happen? Does this say anything about the quality of the molecule?”
Here are some notes from Drug Hunter contributors, including two PharmDs:
Contributor #1: “It’s likely that the clinical data didn’t evolve to be as good as the initial results made it seem (possibly safety related). It’s not an unheard of occurrence for BTDs to be rescinded, and the FDA tallies previously rescinded BTDs online here. It’s not necessarily the molecule’s properties but more likely the clinical data around it. The FDA has new draft guidance on considerations for rescinding BTD here.”
Contributor #2: “[Looking at] disease treatment options for PV and the FDA's definition of Breakthrough Therapy (significant improvement in one or more clinical endpoints compared to standard treatment), it seems like there's probably no previous true comparator/pharmacological agent as a standard treatment. In my opinion, that’s a likely reason the FDA is rescinding it in PV. I'm not sure why CDER gave the Breakthrough Therapy Designation in the first place. It could be the case that it is being rescinded after a senior review at CDER 🙂”
Contributor #3: “It’s very rare for the FDA to rescind BTDs, and the few times they’ve done it, the reasons cited were clinical data. It’s hard to believe that rescinding rusfertide would be related to clinical data since it seems the data from their Ph. II trial looked good. It could be that some new data may have come to light.”
We’d love to hear what readers think!