Computational Partnership in Small Molecule Drug Discovery
Sponsored by:

Schrödinger is transforming the way therapeutics and materials are discovered. Schrödinger has pioneered a digital chemistry software platform that enables discovery of high-quality, novel molecules for drug development more rapidly and at lower cost compared to traditional methods. The platform combines the power of physics-based molecular simulation with machine learning and team-based collaborative informatics. The software is used by biopharmaceutical and industrial companies, academic institutions, and government laboratories around the world.
The real magic happens when chemists can integrate computational models into their assay cascades, thanks to trust in their predictions. To get there, grounding computational predictions in physics and experimental reality makes all the difference.
In this article, Abba E. Leffler, Ph.D. from Schrödinger tells us about:
How to make computational tools like WaterMap and FEP+ work for you
Why physics makes a difference: avoiding garbage in, garbage out
How modeling can lead to non-obvious design ideas
Avoiding synthetic land wars by taking “calculated” risks
Example of computational models in action from the Structure Based COVID Alliance Project
How to Make Computational Tools Work For You
When used within their domain of applicability, computational tools can:
Empower teams with broader sets of better-validated hypotheses to test
Facilitate more confidence in novel designs by de-risking synthesis
Save time and experimental resources with rapid in silico design cycles
To fully unlock the power of computational workflows:
The model predictions should be accurate and trustworthy to chemists
The predictions and data should be accessible to the whole design team
The project team should embed computational tools into standard workflows
When chemists trust their computational predictions and treat computational models as part of the assay cascade, the real magic happens. Rather than handing chemical exploration over to a computational black box, the software becomes a trusted collaborator – even when it provides non-intuitive results that push the program into uncharted chemistry.
Physics Makes a Difference – Avoiding Garbage In, Garbage Out
The importance of trust in computational tools can’t be understated. For chemists to embrace computational predictions, they must deliver routinely reliable prospective value. Poor predictive power could result in chemists losing trust in models, and lead to the workflows that integrate computational models falling apart.
Models built on physics and grounded in well-validated, accurate structural data can have a needle-moving impact on potency and property prediction. Low quality, unreliable, or inappropriate models used in program workflows can be worse than no model at all and run the risk of turning chemists off to any future attempt to bring in other computational tools. As chemists are fond of saying, “garbage in, garbage out.”
Successful computationally-enabled project teams not only excel at understanding the fundamental physics behind how models are constructed but also understand when their use is appropriate for the drug discovery team. Appreciating the domain of applicability of a model and committing to continually working to improve it when new data are available is key for effectively driving a drug discovery program with computational methods.
Water You Looking For? Non-obvious Design Ideas from Modeling
When designing compounds, one should continually ask: “Where should I be looking next?” For example, a computational method that calculates the positions and energies of water sites in a protein binding pocket (WaterMap) can help answer this question by pointing out “where” and “how” to focus design efforts:
Where: If a water site is high-energy or “unstable,” then adding a moiety to the compound that displaces a water molecule from that site should increase its potency. Focusing on targeting those water sites is a good starting point.
How: Water sites come in two flavors: replaceable and displaceable. Replaceable waters should be targeted with a polar group, while displaceable waters should be targeted with a hydrophobic group.

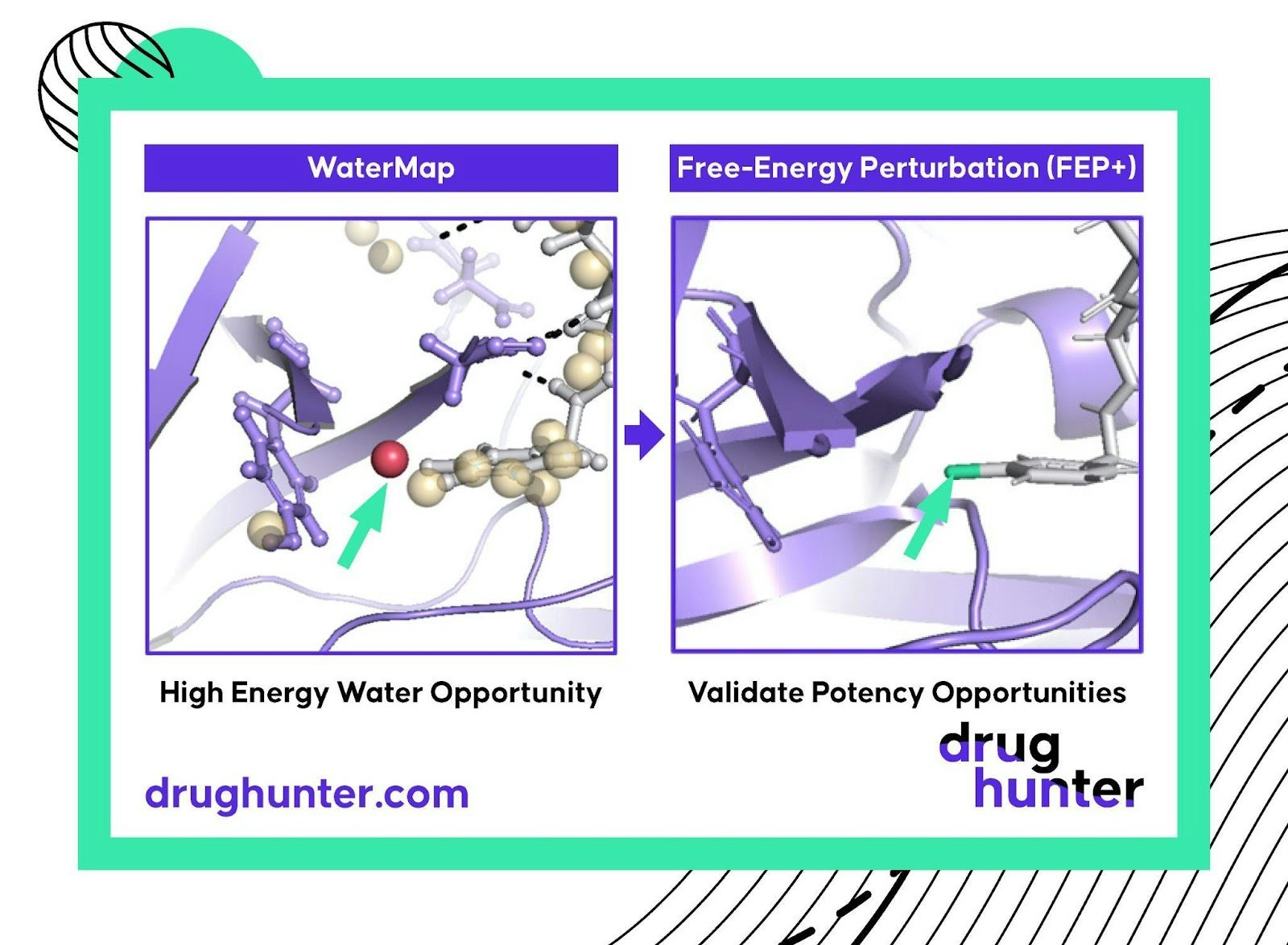
Figure 1. Computational tools like WaterMap can help identify where significant potency gains might be achieved with SAR, while predictive methods like FEP+ can help validate design hypotheses in taking advantage of these opportunities. Example shown above is from a program involving thrombin inhibitors.
Recent Example: Structure Based COVID Alliance Project
A collaborative initiative with scientists from Schrödinger, Takeda, Novartis, Gilead Sciences, and WuXi
While working on the discovery of a SARS-CoV-2 Main Protease (Mpro) inhibitor as part of the recent Structure Based COVID Alliance Project, one of the first things we did was run a WaterMap analysis of the Mpro active site using a high-resolution x-ray structure. I was excited not only to see several high-energy water sites in the Mpro active site pockets but also to observe that existing chemical matter did not target one of those water sites.

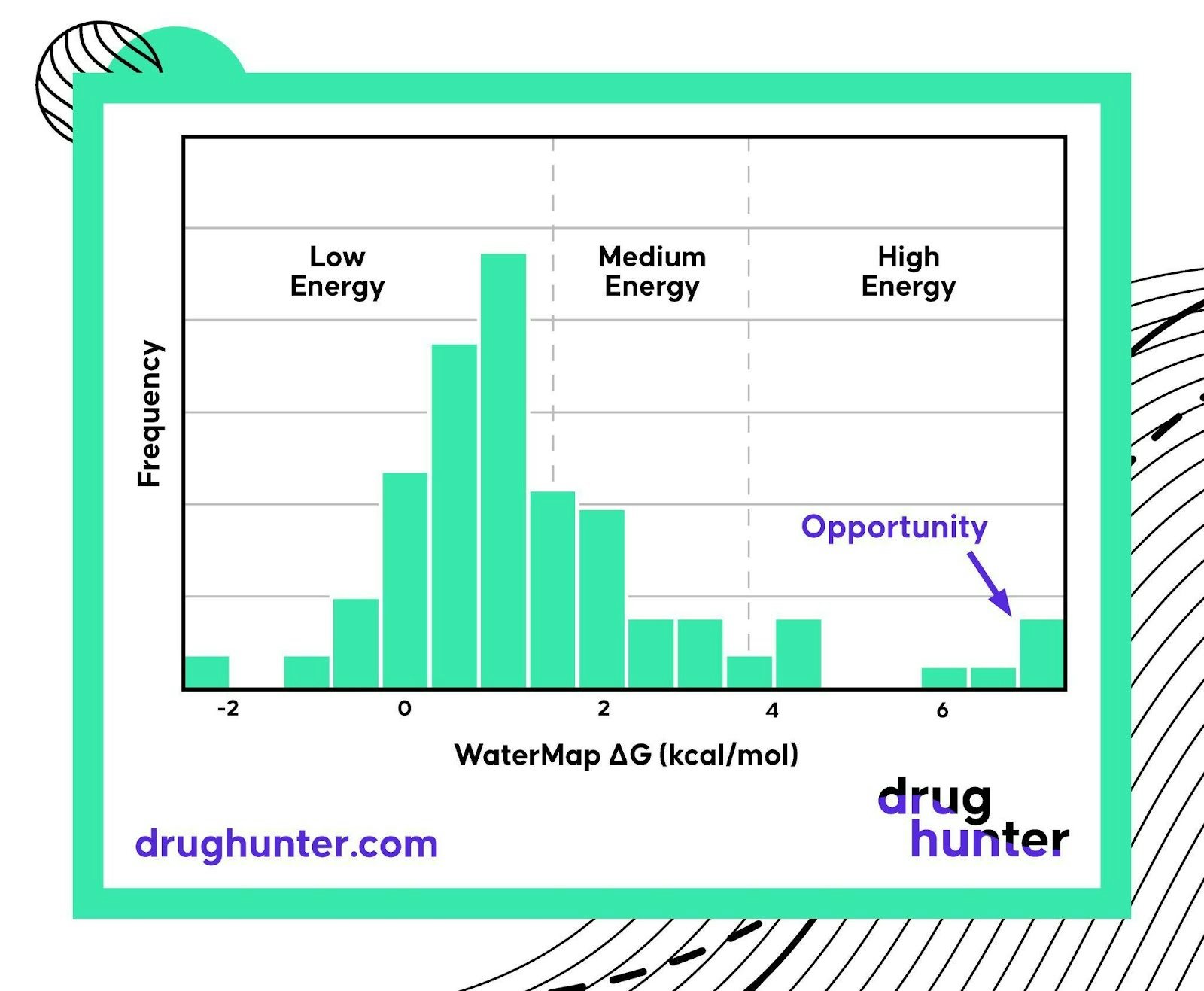
Figure 2. WaterMap can be used to calculate the free energy of water molecules in a protein binding site, helping identify high energy waters that can lead to significant potency gain opportunities if displaced by an appropriate chemical modification.
Our team of medicinal and computational chemists, based in collaborator companies across the globe, worked together to design compounds with R-groups that targeted this water site using the shared collaborative ideation and design platform, LiveDesign. Some of the ideas were creative and non-intuitive because of where this water site was located; we had to explore new vectors and functional groups to ‘pop out’ the desired water from its crevice. This turned out to be worth the effort, as several of these compounds showed enhanced inhibition of Mpro once they were synthesized and assayed, leading to a new design direction for the team.
Avoid the Land War: Taking Calculated Risks with Calculated Free Energies (FEP+)
The more difficult a compound is to make or the more novel it is, the more confidence you want that it’s actually going to be potent for your target. Late in the game, even analogs with single atom changes (such as from a carbon to a nitrogen atom) within the core of a molecule can require the re-design of an entire synthetic route. This is one area where highly rigorous methods like free energy perturbation (FEP+) can really save time and resources - it’s computationally easy to test changes so that you can avoid synthetic “land wars.”
FEP+ is a physics-based simulation method for predicting the change in affinity of hundreds or even thousands of compounds at a receptor relative to reference compounds with known experimental data.
FEP+ is not docking: the receptor and compound motions are sampled using molecular dynamics simulation, waters are present, and the thermodynamic cycle of binding is simulated.
Extensive development of the utilities that support accurate FEP+ calculations, such as on-the-fly fitting of torsional potentials, make it viable to profile the entire synthesis queue so that the most promising compounds can be prioritized.
In addition to on-target potency, FEP+ can help guide off-target selectivity and work alongside other physics-based and machine learning predictions to guide multi-parameter optimization. This allows you to balance key properties like solubility, permeability, and off-target binding, all while maintaining the potency level you need.


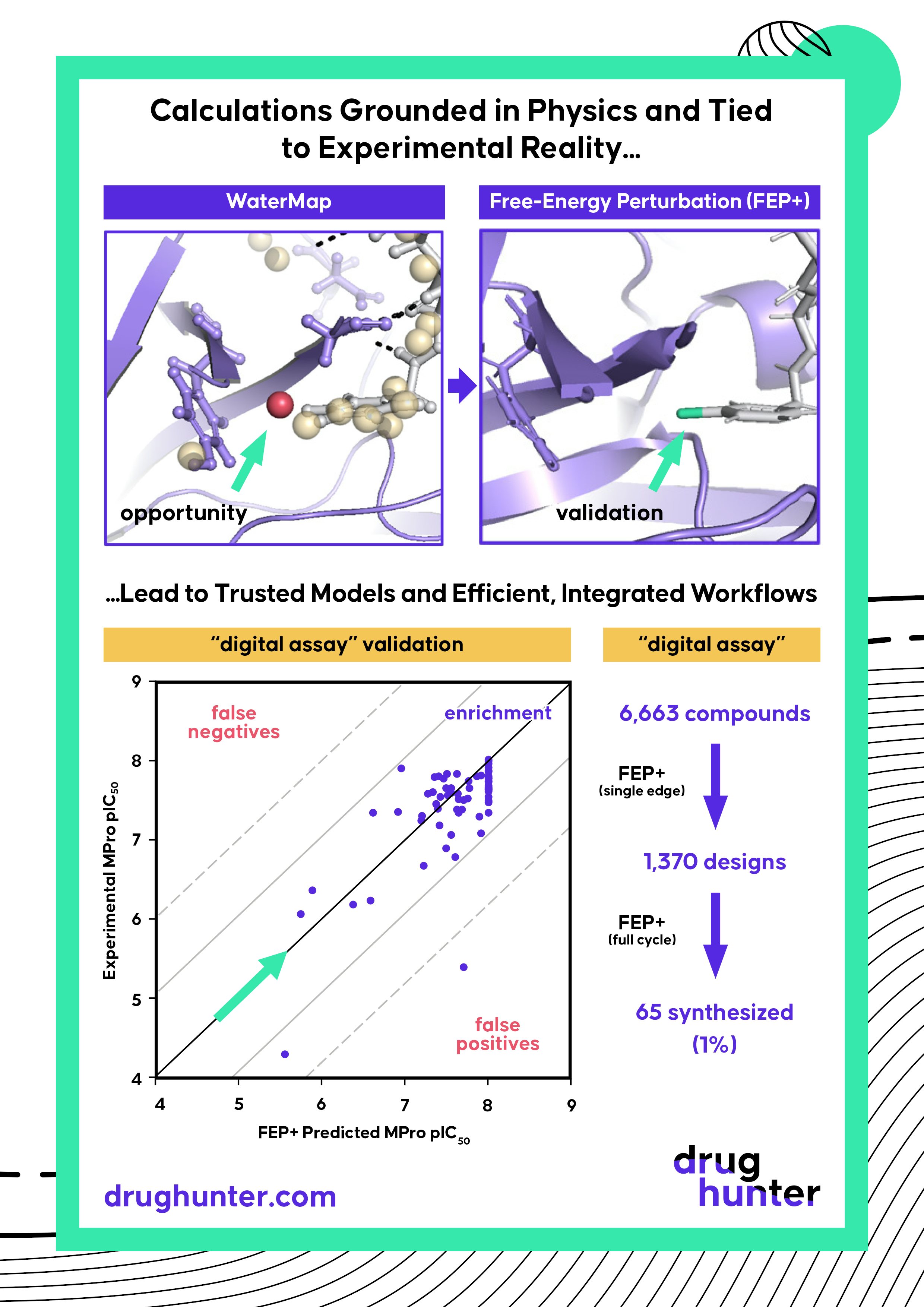
Figure 3. When a computational model has been sufficiently validated, it can become relied on as a “digital assay,” helping to prioritize designs for synthesis. While predictions are not perfect, they can quickly remove designs that have low probability of success. Experimental activity values of compounds against MPro correlated well with calculated activity values using FEP (left), allowing the model to be used to quickly triage designs (right).
Recent Example: Structure Based COVID Alliance Project
Using FEP+ to prioritize compounds for synthesis during the Mpro project gave the team confidence to pursue ideas that ran counter to the existing SAR. It was rewarding when compounds that contained R-groups, unlike those we had observed in the literature, came back as potent as predicted. While 97% of our prospective predictions were within a log unit of the subsequently measured experimental value, more importantly, the high accuracy of FEP+ enabled the project to progress rapidly. Within a few Design-Make-Test-Analyze (DMTA) cycles, we had identified compounds as potent biochemically and in cells as the FDA-approved Mpro inhibitor, PF-07321332 (nirmatrelvir).
Discover Better, Together: Facilitating Collaboration to Drive Efficient, Effective Design Cycles
Platforms that combine access to predictive rigorous computational models with the ability to rapidly generate ideas together across computational and medicinal chemistry teams help drive the discovery of novel compounds with desirable properties more quickly. They allow you to:
Crowdsource design ideas by giving chemistry teams access to powerful design tools and project-specific predictive models
Breakdown communication silos by working across teams and geographies - everyone on the team can see the same information and build off each other’s ideas
Improve the organization of data and extraction of insights by allowing the team to identify the most important areas to focus on quickly and integrate other pharmacology or ADME data.
Recent Example: Structure Based COVID Alliance Project
The accuracy of FEP+ on the Mpro program inspired me to think about ways that we could get it into the hands of medicinal chemists to unleash their creativity. My goal was to enable medicinal chemists to design compounds, run FEP+, and analyze the resulting virtual SAR - all directly in a single collaborative design and ideation platform (LiveDesign).
Over 6,000 FEP+ calculations were run using this approach, and medicinal chemists adopted different styles when using it. One medicinal chemist used an approach that I found ingenious. Every day he would design a cohort of around fifty compounds and then launch FEP+ on them in LiveDesign. When the results finished the next morning, he would design a new cohort of compounds based on this “virtual SAR” and then run FEP+ on the new set. By democratizing FEP+ so that medicinal chemists could run it directly, the team made breakthrough discoveries in record time.
Conclusion
The most effective computational tools are ones that provide predictive accuracy that gains the trust of chemistry teams, thus facilitating the exploration of more creative design ideas and dramatically expanding in silico hypothesis testing. Bringing it all together in a single collaborative enterprise platform can unleash the collective power of teams of scientists in the development of better quality drugs for patients faster.
If you found these case studies inspiring, consider reaching out to our team at Schrödinger to empower your drug discovery programs with a digital chemistry strategy:
You can also gain hands-on experience with Schrödinger computational tools and learn best practices by registering for one of the upcoming courses:
Online Course: Introduction to Molecular Modeling in Drug Discovery
Online Course: High-Throughput Virtual Screening for Hit Identification and Evaluation
Acknowledgements: The author would like to thank Leah Frye, Ph.D., Distinguished Fellow in the Drug Discovery group at Schrödinger, and Daniel Carney, Ph.D., Senior Scientist at Takeda, for their key roles in the Structure Based COVID Alliance Project and for their work on the figures and underlying data that are featured in this article. Additionally, thank you to all the organizations and team members that collaborated on the Structure Based COVID Alliance Project – from Takeda, Novartis, Gilead Sciences, WuXi, and Schrödinger.
Download the poster summary below: