Billion-Dollar Biotransformations: On the Metabolism of Ozanimod
Sponsored by:


Today, ozanimod is a likely megablockbuster drug approved for ulcerative colitis and multiple sclerosis. But before it became the first S1P modulator approved in ulcerative colitis, it received a scary, headline-making “Refuse to File” letter from the FDA due to insufficient preclinical characterization of an active metabolite RP112273 (CC112273). Addressing the refusal to file letter and FDA concerns around potential drug-drug interactions led to a two-year delay before ozanimod was approved and marketed, resulting in likely billions of dollars of lost sales and risking its leading position in the competitive S1P modulator space. The case study highlights the value of intricately understanding drug metabolism early in the drug discovery process. In this article, Dr. Julia Shanu-Wilson of Hypha Discovery walks us through the fascinating metabolism of ozanimod in detail.
Overview of Ozanimod Metabolism
Metabolism of the drug ozanimod (Zeposia®, BMS) must surely take the top prize in biotransformational intrigue. Originally invented by Scripps Research’s scientists, its development passed through Receptos and Celgene before finally winding up under the BMS umbrella. Its approval was delayed by quirks in its metabolism that were discovered late in development, including the presence of more than seven active metabolites that can be as potent and as selective as the parent drug. In fact, a single, accumulating active metabolite, CC112273, has much greater exposure than the parent molecule, accounting for up to 90% of all drug-related exposure at steady state.

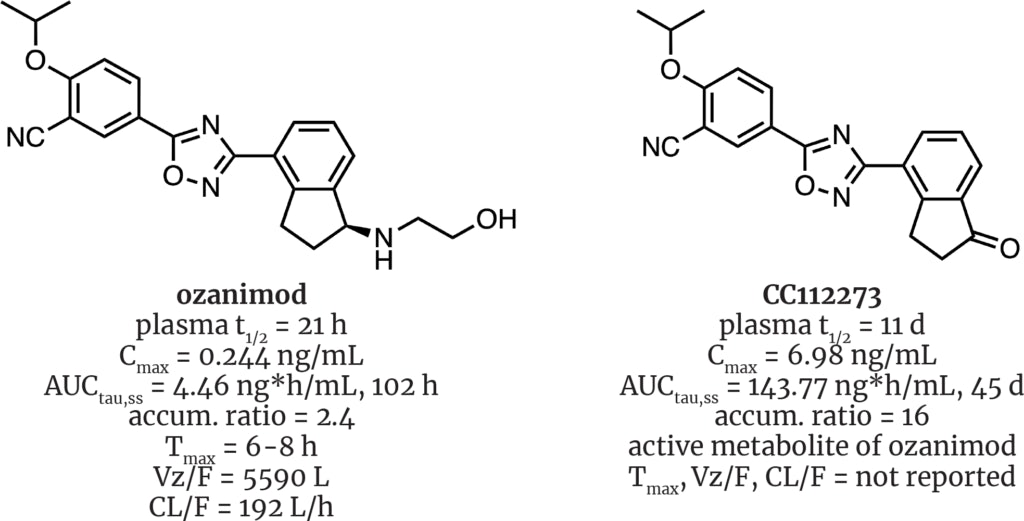
A recent talk by BMS VP of Nonclinical Disposition and Bioanalysis, Sekhar Surapaneni, highlighted the complexity of ozanimod’s metabolism. Not only are familiar suspects involved (CYP3A4/1A1), but other less frequently invoked enzymes such as ADH/ALDH, NAT2, MAO-B, CYP2C8, CBR, and AKR/HSD play important roles.

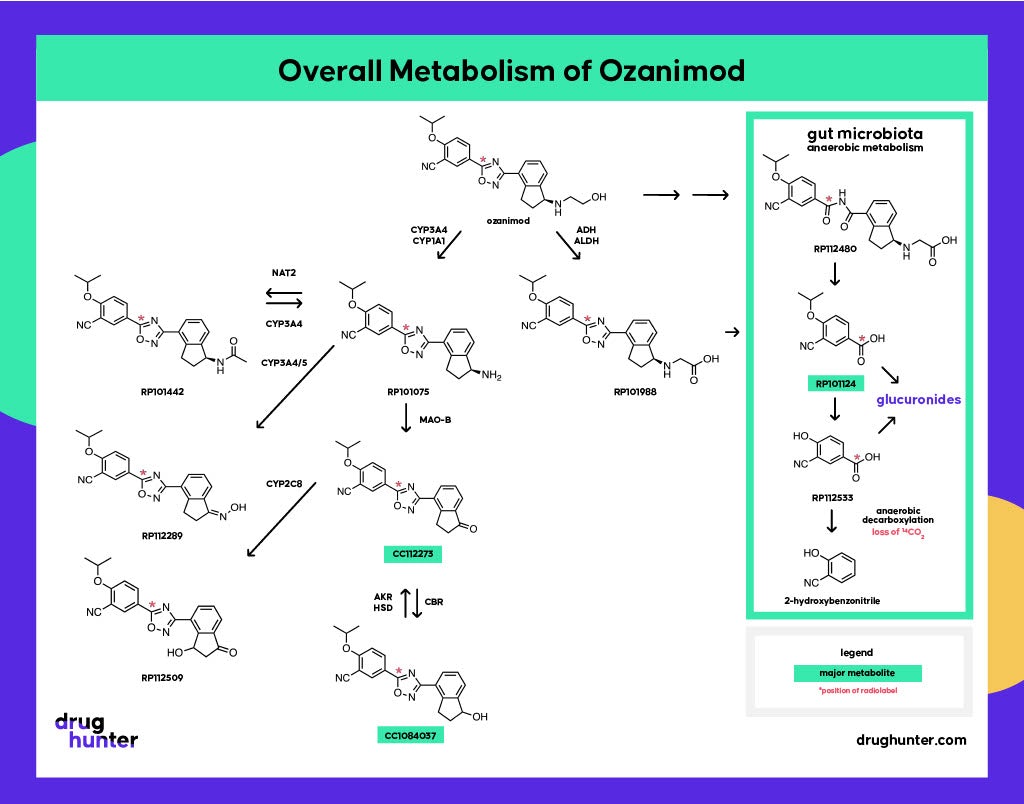
The story contains great lessons, including the importance of drug radiolabeling studies in identifying human metabolites, the role of active metabolites in drug-drug interactions and species differences, the need to account for gut microbial metabolism in DMPK studies, and the underappreciated role of monoamine oxidase enzymes (MAOs) in CYP-like drug metabolism.
Impact of the Human 14C AME Study
The human 14C AME (hAME) study was conducted late in development in parallel to Ph. III studies. hAME studies use radio-labeled versions of a drug to evaluate the pharmacokinetics, mass balance, routes of excretion, and metabolic pathways of the drug to confirm that its metabolic profile is similar to what was observed in preclinical ADME testing. These studies are done to help identify any uniquely human or disproportionate metabolites. In the case of ozanimod, the results raised an alarm bell due to the large % of circulating radioactivity that could not be accounted for by metabolites known at that time. Investigations nailed a new metabolite, CC112273, that proved to be equally potent as ozanimod towards S1P1 and S1P5 and with a much greater overall exposure than the parent drug, driving pharmacology.
If that wasn’t enough, there are additional twists involved in the clearance of CC112273, which impacts its accumulation and, at steady state, ends up as ~90% of all drug-related exposure. CC112273 requires the prior formation of RP101075 (N-dealkylated metabolite), which is then metabolized by MAO-B. CC112273 is then channeled through one of two pathways to RP112509 and CC1084037.

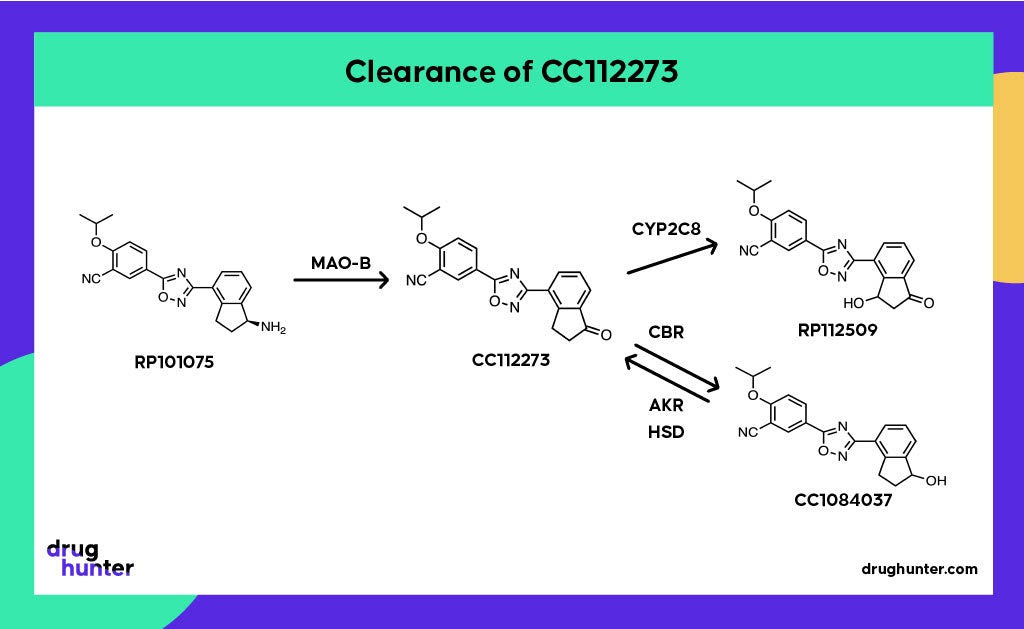
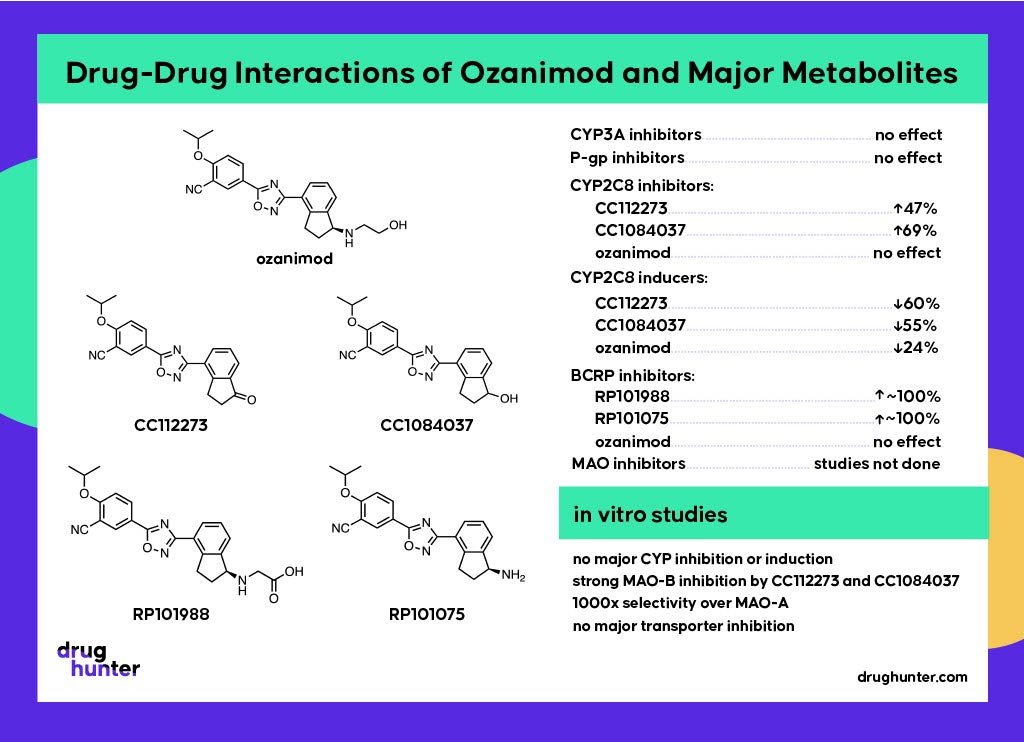
Keto reduction of CC112273 by carbonyl reductase (CBR) forms CC1084037, another major metabolite at steady state. It turns out that these metabolites are interconvertible, with CC1084037 being rapidly oxidized back to CC112273 by multiple enzymes, including aldo-keto reductases (AKRs) and hydroxysteroid dehydrogenases (HSDs). CC112273 predominates because the rate of the reverse oxidation reaction is greater than the rate of reduction, which is contrary to what would expect for oxido-reduction reaction where the reduced metabolite is more polar and hence usually more rapidly cleared. Clearance of CC112273 involves mono-oxidation of the indanone ring system by CYP2C8 to form RP112509; hence drugs that inhibit CYP2C8, such as gemfibrozil and its acyl glucuronide, are contraindicated. As it happens, enhanced clearance of CC112273 and CC1084037 prevails in animals, making these disproportionate human metabolites.

Recently, human-first, human-only ADME strategies have been championed by some big pharma companies, where the drive is to move the identification of metabolites earlier in order to reduce the risk of unwelcome surprises later in development.
The Need to Account for Gut Microbial Metabolism
Gut microbial metabolism is often underappreciated in human drug metabolism. Interestingly, on top of the other unusual features of ozanimod metabolism, there is also the involvement of gut microbiota to generate a major inactive metabolite, RP101124. This metabolite, formed as a result of oxadiazole reduction and hydrolysis of the intermediary amide, was discovered during an in vivo 14C-ADME study in rats, where a lag time was observed before its appearance in circulation.

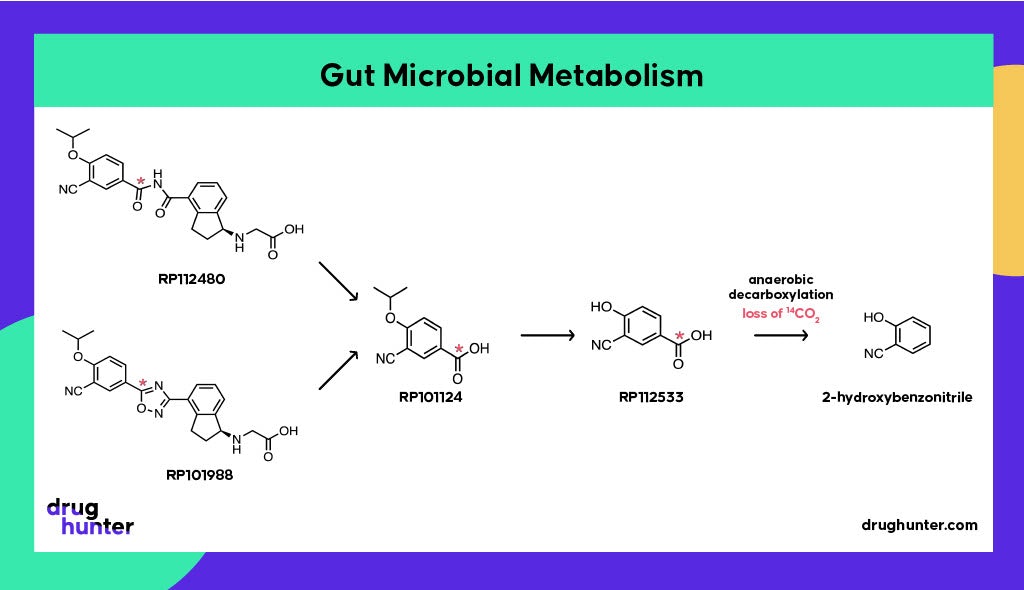
Antibiotic studies revealed the involvement of gut microflora in the formation of RP101124 from both ozanimod and RP101988 under strict anaerobic conditions. Antibiotics do not affect the extent of exposure for ozanimod or its metabolites (Surapaneni, personal communication), although there are multiple examples where antibiotics (and non-antibiotic drugs) cause gut microbial dysbiosis and consequently affect drug metabolism and disposition. However, literature on the impact of antibiotics on gut microflora-mediated reductive metabolism is mixed.
Gut bacteria are also involved in the breakdown of RP101124, via RP112533, through anaerobic decarboxylation; this step may have contributed to the low recovery in the human mass balance study where radiolabel was lost as 14CO2, in addition to the long half-life of metabolites. Only 60% of the expected radioactivity was recovered from urine and feces. This example highlights how the human microbiome can contribute to unexpected drug biotransformations, potentially complicating data interpretation ahead of regulatory filings.
The Underappreciated Role of Monoamine Oxidases in CYP-Like Transformations: MAO-B
A fourth unusual aspect of ozanimod’s disposition is the significant role of a monoamine oxidase (MAO-B) in its metabolism.

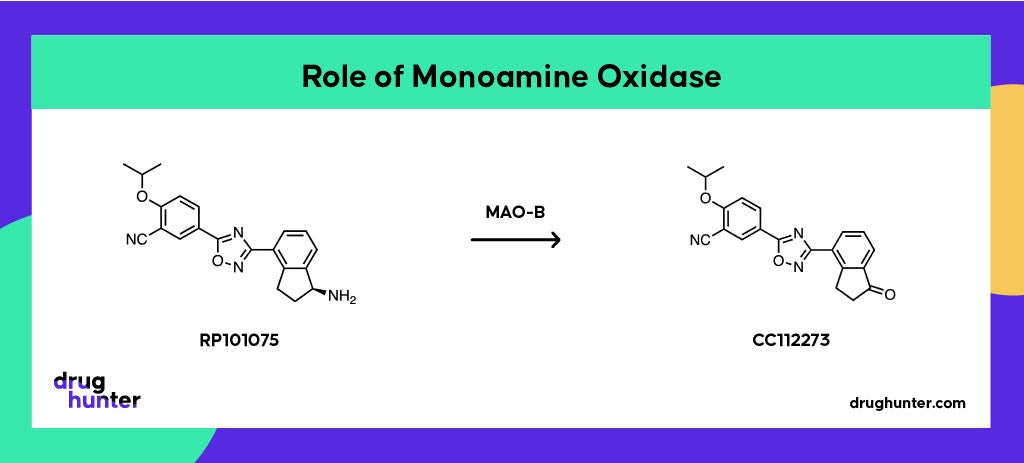
Oxidative deamination of an intermediate metabolite (RP101075) by MAO-B forms a disproportionate active metabolite (CC112273), which turned out to be the main pharmacological driver on account of its long half-life and accumulation at steady state.
Monoamine oxidases catalyze the oxidative deamination of amine-containing compounds, including several neurotransmitters, where rapid degradation of these by MAO isoenzymes is essential for appropriate synaptic neurotransmission. MAOs have distinct but overlapping substrate specificities and are responsible for only ~1% of xenobiotic metabolism. The initial assumption was that the formation of CC112273 was mediated by CYPs. However, the formation of the metabolite in incubations without the addition of NADPH (required for CYP activity) pointed to a different mechanism being at play, even though the CYP inhibitors aminobenzotriazole (ABT) and ketoconazole were shown to inhibit the formation of CC112273. It turns out that these widely used “CYP inhibitors” also inhibit MAO-A and MAO-B!


To distinguish which MAO was involved, experiments using very selective MAO inhibitors clorgyline (MAO-A) and deprenyl (MAO-B) revealed the exclusive involvement of MAO-B. Most drugs that are substrates of MAOs are either metabolized by both isoforms or only MAO-A, making ozanimod somewhat unusual in its MAO-B specificity. This finding also explains why MAO-B inhibitors such as selegiline, phenelzine, and linezolid are contraindicated with ozanimod.
Conclusion
Ozanimod has demonstrated impressive activity in multiple indications, but its approval was nearly derailed by insufficient characterization of an active metabolite. Thanks to the efforts of numerous scientists to understand the drug’s metabolism, ozanimod made it over the line.
There are multiple lessons to be drawn here, including the utility of early radiolabeling studies to identify key human metabolites, the key role of active metabolites in drug-drug interactions and species differences, the importance of the human microbiome in drug metabolism, and the importance of MAO activity in the metabolism of drugs. I recommend reading the papers in the reference list to appreciate the details surrounding the discovery of all the metabolic routes related to ozanimod.
I hope the metabolism of ozanimod was as fascinating to you as it was to us at Hypha Discovery, where we specialize in making, purifying, and characterizing drug metabolites. If your team ever needs support in this area, please reach out to us here.
Glossary of significant non-CYP enzymes involved in the metabolism of ozanimod in humans
AKR: aldo-keto reductase
ALDH/ADH: aldehyde dehydrogenase and alcohol dehydrogenase
CBR: carbonyl reductase
HSD: hydroxysteroid dehydrogenase
MAO-B: Monoamine oxidase B
NAT2: N-acetyltransferase 2
Acknowledgments
Julia would like to acknowledge Sekhar Surapaneni’s review of the information included in this article and for additional useful comments about the gut metabolism of ozanimod.
References
April Bai, Veerabahu Shanmugasundaram, Julie V. Selkirk, Sekhar Surapaneni and Deepak Dalvie. MAO B–Mediated Metabolism of Ozanimod Metabolite RP101075. Drug Metabolism and Disposition August 1, 2021, 49 (8) 601-609.
Ma, Y., Liu, X., Wang, J. Small molecules in the big picture of gut microbiome host cross-talk. The Lancet, 104085, July, 2022.
Rendić, S.P., Crouch, R.D. & Guengerich, F.P. Roles of selected non-P450 human oxidoreductase enzymes in protective and toxic effects of chemicals: review and compilation of reactions. Arch. Toxico.l 96, 2145–2246 (2022).
Sekhar Surapaneni, Talk on Ozanimod Metabolism – Qualification of Disproportionate Metabolites (ISSX webinar 11th April 2020).
Sekhar Surapaneni, Usha Yerramilli, April Bai, Deepak Dalvie, Jennifer Brooks, Xiaomin Wang, Julie V. Selkirk, Yingzhuo Grace Yan, Peijin Zhang, Richard Hargreaves, Gondi Kumar, Maria Palmisano and Jonathan Q. Tran. Ozanimod Human Metabolism and Disposition. Drug Metabolism and Disposition May 1, 2021, 49 (5) 405-419.
Tran JQ, Zhang P, Walker S, Ghosh A, Syto M, Wang X, Harris S, Palmisano M. Multiple-Dose Pharmacokinetics of Ozanimod and its Major Active Metabolites and the Pharmacodynamic and Pharmacokinetic Interactions with Pseudoephedrine, a Sympathomimetic Agent, in Healthy Subjects. Adv. Ther. 2020 Dec;37(12):4944-4958.
Young, G.C., Spracklin, D.K., James, A.D., Hvenegaard, M.G., Scarfe, G., Wagner, D.S., Georgi, K., Schieferstein, H., Bjornsdottir, I., van Groen, B., Romeo, A.A., Cassidy, K.C., Da-violante, G., Bister, B., Blech, S., Lyer, R., Schulz, S.I., Cuyckens, F. and Moliner, P. (2022), Considerations for Human ADME Strategy and Design Paradigm Shift(s) – An Industry White Paper. Clin. Pharmacol. Ther.