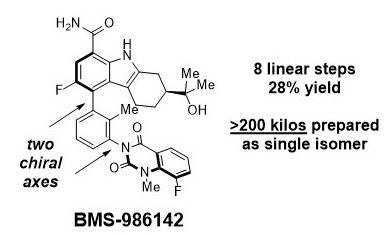
Adventures in Atropisomerism: A Case Study from BMS
BMS-986142 is a reversible BTK inhibitor with two axial stereocenters. BMS synthetic chemists were able to make >200 kilos of this compound as a single isomer using this route. Wow.
Atropisomers are a nightmare in drug discovery. Axial stereocenters are not easy to set predictably, separation of isomers is a pain, and the products are hard to assign. The solution for many medicinal chemists is to avoid making axially chiral molecules altogether. But sometimes you have no choice, as was the case in BMS's reversible BTK inhibitor program. [(a) Watterson, S. H. et. al. J. Med. Chem. 2016, 59, 9173-9200. (b) DeLucca, G. V. et. al. J. Med. Chem. 2016, 59, 7915-7935.] BMS-986142, a lead compound, ended up possessing not one but two chiral axes. Fortunately, BMS has a world-class Chemical & Synthetic Development group, which just published a route used to prepare >200 kilos of this compound as a single isomer. (Beutner, G. et al. Org. Lett. 2018, 20, 3736-3740.)

Building the first stereocenter
The synthesis begins with a rhodium-catalyzed asymmetric conjugate addition of isopropenylboronic acid ester [A] to cyclohexenone [B]. (Lalic, G.; Corey, E. J. Tetrahedron Lett. 2008, 49, 4894) This step was carefully optimized at BMS to employ only 0.3 mol% of rhodium precatalyst while delivering cyclohexanone [C] in 89% yield and >99.5% ee on up to 100 kg scales. (Simmons, E. M., et al. Org. Process Res. Dev. 2017, 21, 1659-1667.) Hydrazone formation, followed by Zinc-mediated Fisher indolization afforded [D] contaminated with the minor regioisomer, which was removed by crystallization from acetonitrile. The olefin in [E] was then converted to alcohol [F] without isomerization via a two-step procedure involving an intermediate TFA-ester.

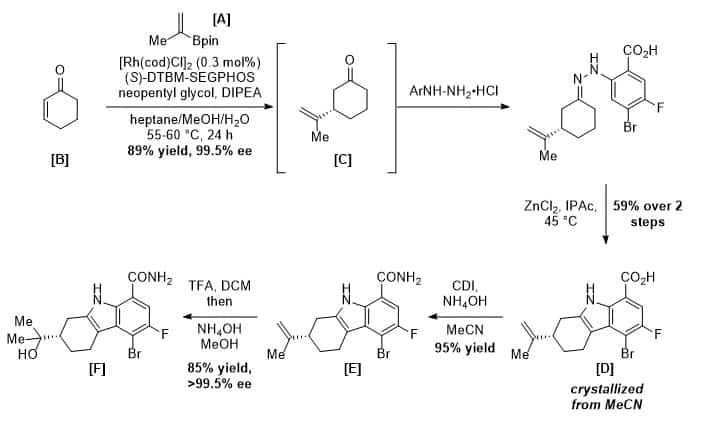
Scheme 1. Synthesis of a chiral indole intermediate.
Constructing the first chiral axis The first, higher-energy barrier chirality axis was set by coupling [F] with aniline [G]. Coupling mediated by achiral catalyst Pd(dppf)Cl2 showed minimal influence by the remote alcohol stereocenter, resulting in an atropdiastereomeric ratio of 1.4 to 1. Though chiral ligand screening showed improvements in dr using (S)-Xyl-SDP and (S)-Ph-SDP, these ligands are extremely expensive on manufacturing scale. Thus the team decided to optimize the reaction using (R)-BINAP instead. By lowering the reaction temperature to 5 ºC and including a MeOH cosolvent to improve boronic acid solubility, product [H] was formed in 16:1 dr. Crystallization from n-BuOH resulted in isolation of [H] in 87% yield with a 65:1 dr.

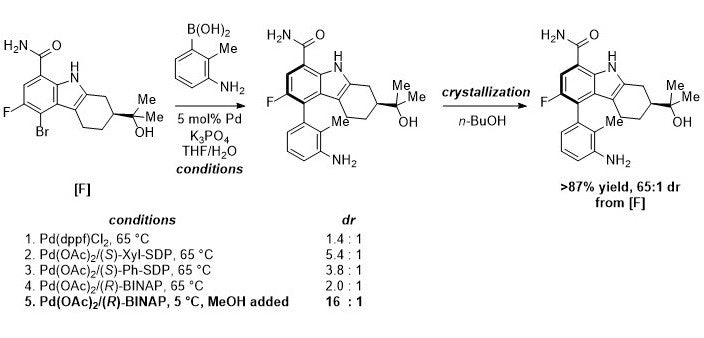
Scheme 2. Highlights from optimization of the first intermediate with axial chirality.
Forming the second chiral axis To form the second axis, the BMS team coupled aniline [H] to acids bearing different carbamates. These carbamates could then be cyclized under basic conditions to afford BMS-986142 [G] as a mixture of atropdiastereomers. While the cyclization of phenylcarbamate [I] resulted in a 1 to 1.5 ratio favoring the undesired diastereomer, the cyclization of n-propylcarbamate [J] resulted in a ratio of 6 to 1 favoring the correct isomer. The selectivity was further improved to 96 to 4 by employing LiOt-Bu as the base in dioxane solvent. Crystallization from a mixture of MeTHF, MeOH, and acetone resulted in the isolation of [G] in 90% yield with <0.5% of the undesired atropisomer.

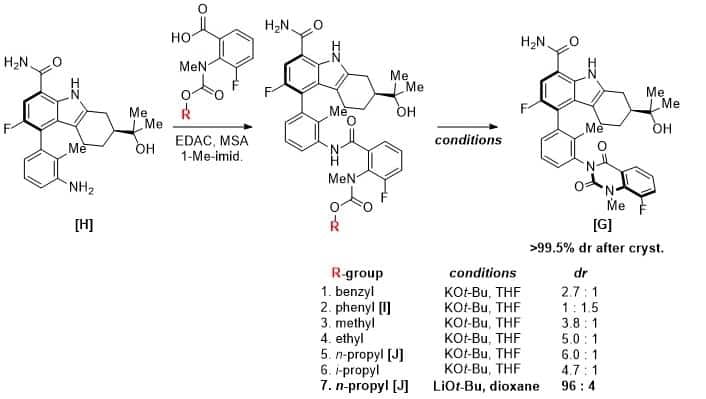
Scheme 3. Highlights from the optimization of the construction of the second chiral axis.
Summary In summary, the BMS team developed an enantioselective and diastereoselective synthesis of BMS-986142 in eight steps and 28.3% overall yield, which enabled the manufacture of >200 kg of [G] to support clinical trials in rheumatoid arthritis. This is a particularly impressive accomplishment given the notoriety of axially chiral molecules in drug design and development. Many thanks to the BMS Chemical & Synthetic Development group for sharing this work with the community in a very readable article! Explore drughunter.com for more.