A Trick to Fluorinate Grignards
Knochel's group has shown that you can fluorinate Grignard reagents without decomposition via single-electron transfer reactivity (SET) by a simple solvent switch from THF to DCM.
A common way to prepare aryl fluorides is through lithium-halogen exchange, followed by trapping with N-fluorosulfonimide (NFSI). This isn't always viable, as the alkyllithium reagents used in the exchange can react with functional groups like nitriles or esters. Magnesium-halogen exchange is often more practical, and Paul Knochel's research group has shown that with in the presence of lithium chloride, Grignard reagents can even be generated in the presence of sensitive functional groups as with ester [A] below. (Knochel, P. et al, Angew. Chemie. Int. Ed. 2003, 42, 4302-4320.) Unfortunately, Grignard reagents aren't as easily fluorinated as aryllithiums. Treating Grignards like [A] with NFSI in THF results primarily in non-fluorinated byproducts, due to single-electron transfer chemistry.

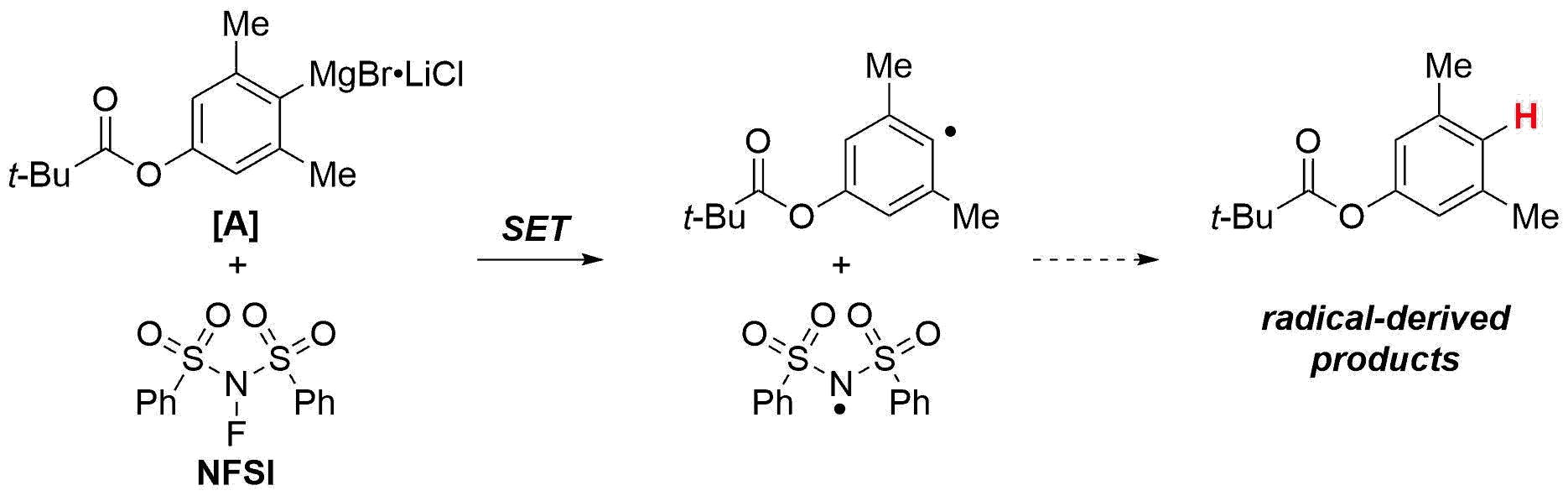
Treatment of Grignard reagents with electrophilic fluorine reagents often does not lead to efficient fluorination due to single-electron transfer (SET) reactivity.
Shigeyuki Yamada and Andrei Gavryushin from Paul Knochel's group in Muenchen discovered a nice, simple solution for this on scales useful for medicinal chemistry. (Yamada, S., Gavryushin, A., Knochel, P., Angew. Chem. Int. Ed. 2010, 49, 2215 –2218)
By replacing the standard THF solvent with dichloromethane after the magnesium-halogen exchange, they are able to fluorinate the arylmagnesium in much higher (>60%) yield. They explain that the DCM solvent helps suppress the byproduct resulting from radical hydrogen abstraction, and that further addition of a fluorinated solvent like perfluorodecalin (A surprisingly inexpensive co-solvent. The reaction works without the perfluorodecalin in slightly lower yield.) can boost the fluorination efficiency further.


By removing THF from the Grignard reagent and re-suspending it in DCM before trapping with NFSI, practical yields of the desired aryl fluoride products can be obtained.
This is a nice, practical trick. I've tried it, and it works! Obviously you should exercise caution when doing solvent switches with Grignard reagents, taking special care to limit exposure to air. On a gram-scale, I've been successful just using a rotovap to do the solvent switch, but on smaller scales and with more reactive reagents you will likely want to try this with inert atmosphere. Doing a solvent switch wouldn't have been the first thing I would have tried when encountering this problem, so kudos to Knochel's team for sorting this out and sharing it with the community. Hope you all also find this useful.
Experimental Procedure

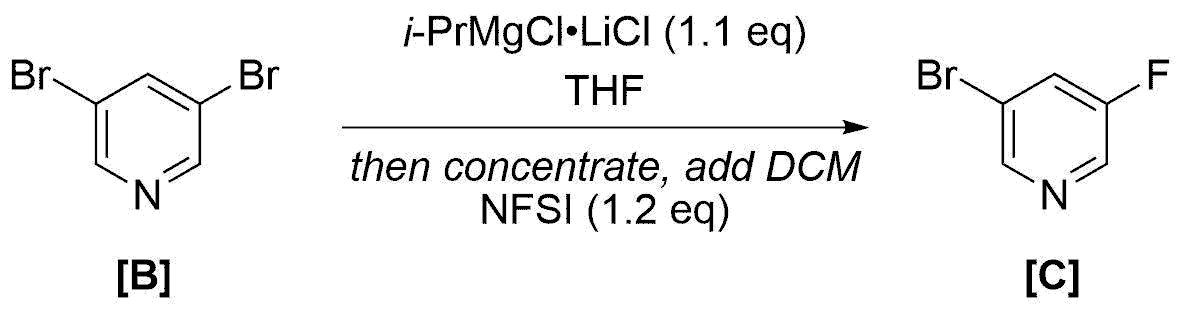
A 50 mL Schlenk flask under N2 was charged with 3,5-dibromopyridine ([B], 1.21 g, 5.0 mmol) in THF (5.0 mL). i-PrMgCl·LiCl (5.5 mmol) in THF (1.16M, 4.7 mL) was added at 0°C and the mixture was stirred at this temperature for 1 h. Then the solvent was removed in vacuo (0.5 mbar, 40°C, 0.5 h). CH2Cl2 (5 mL) was added, and NFSI (1.95 g, 6.0 mmol) in CH2Cl2 (15 mL) and perfluorodecalin (5 mL) was slowly added at -78°C. The reaction mixture was stirred at 0°C for 30 min, then at 25°C for 2 h, and was poured into ice-cooled saturated aqueous NH4Cl (50 mL). After extraction with CH2Cl2 (3 x 50 mL), the organic layers were dried (Na2SO4), filtered, and concentrated in vacuo. The crude residue was purified by column chromatography (SiO2) using pentane/Et2O (20:1) as an eluent, affording [C] (574 mg, 58% yield). The analytical data for [C] are in accordance with those of the commercially available compound.
Explore drughunter.com for more.